シベクトロ®に関する過去のお知らせ一覧
過去のお知らせ一覧

本試験の対象には一部国内承認外の適応菌種が含まれるデータで評価され承認されたため、国内で承認されている効能又は効果と異なるデータも紹介しています。
承認時評価資料:急性細菌性皮膚・皮膚組織感染症患者を対象とした第Ⅲ相試験(ESTABLISH-1;TR701-112試験)
Prokocimer P, et al., JAMA. 2013;309(6):559-569.
[利益相反:本試験は、Trius Therapeutics社(現MSD)が資金を提供し実施した。
P.P.、DA.C.、F.E.はTrius Therapeutics社(現MSD)の株式/ストック オプションを保有し、本研究および分析が行われた時点でTrius Therapeutics社(現MSD)の社員であった。D.A.はTrius Therapeutics社(現MSD)から、コンサルティング料、旅費のサポート、レビュー活動への参加料、原稿料またはレビューに対する報酬を受領し、キュビスト社(現MSD)のコンサルタントである。]
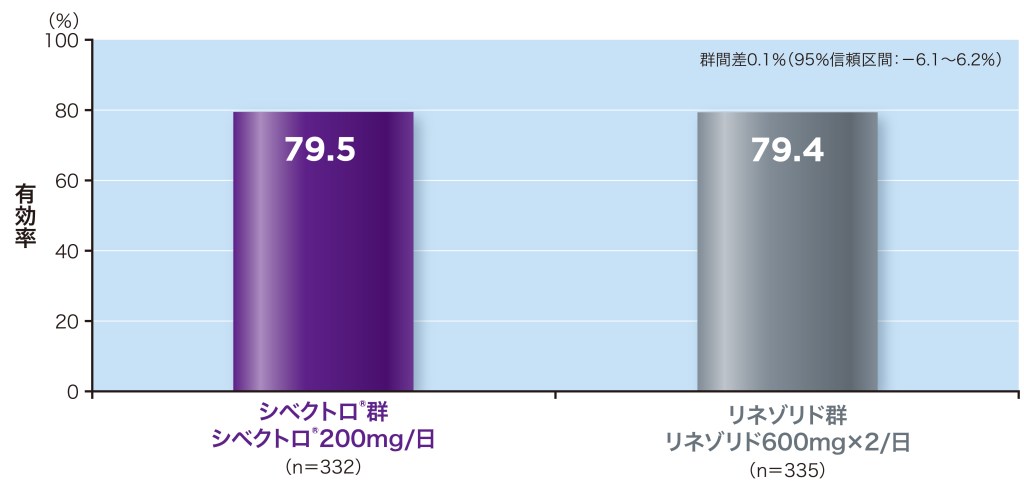
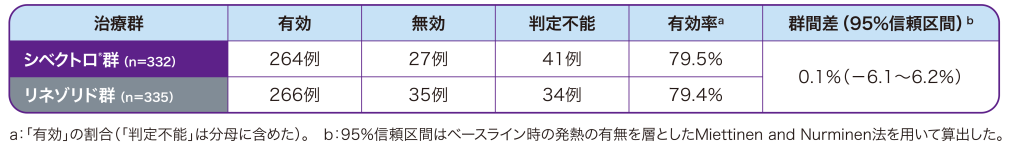
投与開始後48~72時間の早期臨床効果(有効率)は、シベクトロ®群(6日間投与)79.5%、リネゾリド群(10日間投与)79.4%であり、シベクトロ®群はリネゾリド群に対して非劣性であることが検証されました。
以下の基準に基づき、臨床効果をプログラム判定した。
| 判定 | 判定基準 |
| 有効 | 以下の両方の基準を満たした場合 ● 急性細菌性皮膚・皮膚組織感染症の主病変のベースラインからの拡大が停止[病変面積(長さ×幅)がベースラインから増加していない] ● 治験薬投与開始後48~72時間[治験責任(分担)医師が測定]及びその後24時間以内に測定した口腔体温がいずれも37.6℃以下 |
| 無効 | 以下のいずれかの基準を満たした場合 ● 急性細菌性皮膚・皮膚組織感染症の主病変のベースラインからの拡大[病変面積(長さ×幅)がベースラインから増加] ● ベースライン時の原因菌に有効とされる全身性抗菌薬(創感染患者でのアズトレオナム又はメトロニダゾールによる補助投与を除く)を併用 ● 死亡(原因は問わない) ● 治験薬投与開始後48~72時間[治験責任(分担)医師が測定]又はその後24時間以内に測定した口腔体温のいずれかが37.6℃超 |
副作用は、シベクトロ®群331例中80例(24.2%)、リネゾリド群335例中104例(31.0%)に認められました。
主な副作用(発現率2%以上)は、シベクトロ®群で悪心(25例、7.6%)、頭痛(16例、4.8%)、下痢(14例、4.2%)、浮動性めまい(7例、2.1%)、リネゾリド群で悪心(38例、11.3%)、嘔吐(18例、5.4%)、下痢(16例、4.8%)、頭痛(13例、3.9%)、浮動性めまい(7例、2.1%)でした。
重篤な副作用はシベクトロ®群の第7脳神経麻痺、リネゾリド群の自然流産のそれぞれ1例でした。
シベクトロ®群の1例が投与終了後のフォローアップ期間に肺炎、心停止、敗血症性ショックにより第56日に死亡しました(治験薬との因果関係なし)。
投与中止に至った有害事象は、シベクトロ®群で2例[下痢1例、骨髄炎1例](0.6%)、リネゾリド群で2例[悪心2例、嘔吐2例](0.6%)に認められました。®副作用は、シベクトロ®群331例中80例(24.2%)、リネゾリド群335例中104例(31.0%)に認められました。
主な副作用(発現率2%以上)は、シベクトロ®群で悪心(25例、7.6%)、頭痛(16例、4.8%)、下痢(14例、4.2%)、浮動性めまい(7例、2.1%)、リネゾリド群で悪心(38例、11.3%)、嘔吐(18例、5.4%)、下痢(16例、4.8%)、頭痛(13例、3.9%)、浮動性めまい(7例、2.1%)でした。
重篤な副作用はシベクトロ®群の第7脳神経麻痺、リネゾリド群の自然流産のそれぞれ1例でした。
シベクトロ®群の1例が投与終了後のフォローアップ期間に肺炎、心停止、敗血症性ショックにより第56日に死亡しました(治験薬との因果関係なし)。
投与中止に至った有害事象は、シベクトロ®群で2例[下痢1例、骨髄炎1例](0.6%)、リネゾリド群で2例[悪心2例、嘔吐2例](0.6%)に認められました。
試験:国際共同、リネゾリド対照、無作為化、二重盲検、ダブルダミー比較試験(非劣性試験、検証試験)
目的:18歳以上のグラム陽性菌によるあるいはその疑いがある急性細菌性皮膚・皮膚組織感染症患者を対象として、シベクトロⓇ6日間経口投与のリネゾリド10日間経口投与に対する非劣性を、投与開始後48~72時間のプログラム判定による早期臨床効果(有効率、ITT※1にて評価)について検証する。
対象:18歳以上の急性細菌性皮膚・皮膚組織感染症患者(ベースライン時のグラム染色又は培養の結果、グラム陽性菌による感染が疑われる又は確定したもの)[ITT解析対象例数:667例(シベクトロ®群332例、リネゾリド群335例)]
方法:対象患者を、シベクトロ®群又はリネゾリド群に1:1の比で無作為割り付けし、シベクトロ®群にはシベクトロ®
200mgを1日1回6日間、リネゾリド群にはリネゾリド600mgを1日2回10日間、ダブルダミー法にてそれぞれ経口投与した。
評価項目:〈主要評価項目〉投与開始後48~72時間のプログラム判定による早期臨床効果(有効率、ITTにて評価)[検証的解析項目]
〈副次評価項目〉投与終了(EOT)時(第11日)のプログラム判定による臨床効果(治癒率)、投与終了後評価(PTE)時(EOT後7~14日)の治験責任(分担)医師評価による臨床効果(治癒率)、投与開始後48~72時間及び第7日の治験責任(分担)医師評価による臨床効果、各評価時点の疼痛スコアのベースラインからの変化、病変の大きさ、局所徴候及び症状、並びに全身徴候のベースラインからの変化
〈安全性の評価項目〉有害事象、臨床検査、バイタルサイン(血圧、心拍数、呼吸数、体温)、ECG、理学的検査
解析計画:有効性の主要評価項目及び副次評価項目における臨床効果では、有効率(「有効」の割合)について、群間差(シベクトロ®群-リネゾリド群)の、ベースライン時の発熱の有無(無作為化層別因子)で調整した両側95%信頼区間を算出した。また、第1種の過誤の確率を制御するため、Westfall and Krishenの逐次的検定手順を使用し、主要評価項目についてシベクトロ®群の非劣性が検証された場合、副次評価項目について事前に規定した順序で非劣性の検定を行うこととした。群間差(シベクトロ®群-リネゾリド群)の両側95%信頼区間の下限値が-10%より大きかった場合、リネゾリドに対するシベクトロ®の非劣性が成り立つとした。また、MRSA患者の部分集団解析は事前に計画されていなかったが、PTE時の治験責任(分担)医師による臨床効果の検討を行い、評価資料として評価された。安全性の解析は、安全性解析対象集団(SAF※2)を対象とした。
| 評価項目: | 〈主要評価項目〉投与開始後48~72時間のプログラム判定による早期臨床効果(有効率、ITTにて評価) 〈副次評価項目〉投与終了(EOT)時(第11日)のプログラム判定による臨床効果(治癒率)、投与終了後評価(PTE)時(EOT後7~14日)の治験責任(分担)医師評価による臨床効果 (治癒率)、投与開始後48~72時間及び第7日の治験責任(分担)医師評価による臨床効果、各評価時点の疼痛スコアのベースラインからの変化、病変の大きさ、局所徴候及び症状、並びに全身徴候のベースラインからの変化 〈安全性の評価項目〉有害事象、臨床検査、バイタルサイン(血圧、心拍数、呼吸数、体温)、ECG、理学的検査 |
| 解析計画: | 有効性の主要評価項目及び副次評価項目における臨床効果では、有効率(「有効」の割合)について、群間差(シベクトロ®群-リネゾリド群)の、ベースライン時の発熱の有無(無作為化層別因子)で調整した両側95%信頼区間を算出した。また、第1種の過誤の確率を制御するため、Westfall and Krishenの逐次的検定手順を使用し、主要評価項目についてシベクトロ®群の非劣性が示された場合、副次評価項目について事前に規定した順序で非劣性の検定を行うこととした。群間差(シベクトロ®群-リネゾリド群)の両側95%信頼区間の下限値が-10%より大きかった場合、リネゾリドに対するシベクトロ®の非劣性が成り立つとした。また、MRSA患者の部分集団解析は事前に計画されていなかったが、PTE時の治験責任(分担)医師による臨床効果の検討を行い、評価資料として評価された。安全性の解析は、安全性解析対象集団(SAF※2)を対象とした。 |
※1 ITT:割り付けられた全ての患者
※2 SAF:ITTのうち、割り付け後に治験薬を少なくとも1回以上投与された患者
シベクトロ®の4.効能又は効果
< 適応菌種>テジゾリドに感性のメチシリン耐性黄色ブドウ球菌(MRSA)
<適応症>深在性皮膚感染症、慢性膿皮症、外傷・熱傷及び手術創等の二次感染、びらん・潰瘍の二次感染
シベクトロ®の6.用法及び用量
(シベクトロ®錠200mg)通常、成人にはテジゾリドリン酸エステルとして200mgを1日1回経口投与する。
(シベクトロ®点滴静注用200mg) 通常、成人にはテジゾリドリン酸エステルとして200mgを1日1回、1時間かけて点滴静注する。
リネゾリドの4.効能又は効果(詳細は製品電子添文をご参照ください。)
<適応菌種>本剤に感性のメチシリン耐性黄色ブドウ球菌(MRSA)
<適応症>敗血症、深在性皮膚感染症、慢性膿皮症、外傷・熱傷及び手術創等の二次感染、肺炎
<適応菌種>本剤に感性のバンコマイシン耐性エンテロコッカス・フェシウム
<適応症>各種感染症
このサイトでは、医療用医薬品を適正にご使用いただくため、医師、歯科医師及び薬剤師などの医療関係者の方を対象に、主としてMSD株式会社の医療用医薬品に関する情報を提供しています。
下記の「はい」をクリックした場合、「MSD Connect ご利用規約」及び「ウェブサイトのご利用条件」を理解したうえで、内容に同意したものとみなします。
2024年11月にご利用規約を改訂致しました。上記リンクよりご確認ください。
あなたは医療関係者ですか?