成人<同種造血幹細胞移植> 移植後約100日目まで投与 第Ⅲ相国際共同試験(001試験)
臨床成績
成人<同種造血幹細胞移植>
移植後約100日目まで投与
第Ⅲ相国際共同試験(001試験)1、2)
CMV抗体陽性の成人同種造血幹細胞移植患者を対象に臨床的に意味のあるCMV感染の予防*を目的としてプレバイミス®を投与した際の安全性及び有効性を評価する二重盲検無作為化プラセボ対照第Ⅲ相試験(検証試験)
1)承認時評価資料(第Ⅲ相国際共同試験:001試験)
2)Marty FM et al. N Engl J Med. 2017; 377(25): 2433-2444. 利益相反:本試験はMSDより資金提供を受けている。
*:プレバイミス®の効能又は効果は、「下記におけるサイトメガロウイルス感染症の発症抑制 〇同種造血幹細胞移植 〇臓器移植」である。
試験概要
主要目的:
プレバイミス®又はプラセボを投与した際の、移植後24週(約6ヵ月)以内の臨床的に意味のあるCMV感染の予防に対するプレバイミス®の有効性を検討する。
対象:
CMV抗体陽性の成人同種造血幹細胞移植(HSCT)患者570例(うち日本人は36例)
試験デザイン:
国際多施設共同無作為化二重盲検プラセボ対照比較試験[検証試験]
投与方法:
対象を治験実施医療機関及びCMV感染のリスク因子(高リスク/低リスク)別に層別して2:1の比でプレバイミス®群又はプラセボ群のいずれかに無作為に割り付け、移植日から移植後28日までの期間内に、プレバイミス®480mg(シクロスポリン併用時には240mg)又はプラセボの投与を開始し、1日1回経口又は静脈内投与にて移植後14週(約100日)まで継続した。
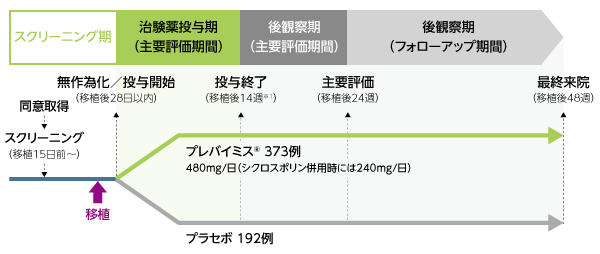
※1:投与期間は14週間ではなく、移植後14週目まで投与(投与期間は開始時期によって異なる)
評価項目:
【主要評価項目】移植後24週以内に臨床的に意味のあるCMV感染※2がみられた患者の割合(検証的解析項目)
【副次評価項目】移植後14週以内に臨床的に意味のあるCMV感染がみられた患者の割合
移植後24週以内に臨床的に意味のあるCMV感染がみられるまでの期間
(移植日から臓器障害を伴うCMV感染症の発症日又は抗CMV薬による先制治療の開始日までの日数) 等
【探索的評価項目】移植後48週以内にCMV感染症がみられた患者の割合
移植後14週、24週及び48週以内に死亡した患者の割合(死亡の原因は問わない)
移植後14週及び24週以内の生着※3率及び生着までの期間 等
※2:臨床的に意味のあるCMV感染は以下のように定義した。
「臓器障害を伴うCMV感染症の発症」もしくは「先制治療の開始(中央測定機関でPCR法によりCMV血症が確認された場合及び患者の状態に基づく)」
※3:3日連続して好中球絶対数が500/mm3以上の場合と定義した。
【安全性】有害事象、臨床検査(血液学的検査、血液生化学的検査、尿検査)、バイタルサイン、12誘導心電図 等
解析計画:
- 有効性の主要解析対象集団は、無作為割付け後に治験薬の投与を1回以上受け、ベースライン時点でCMV DNAが検出されないすべての患者から構成される最大の解析対象集団[FAS(Full analysis set)]とした。
- 有効性の主要解析は、層別因子のCMV感染リスク因子(高リスク/低リスク)で調整したMantel-Haenszel法により群間差を算出し、片側p値が0.0249以下の場合に、プラセボ群に対するプレバイミス®群の有効性が示されたとした。また、欠測値(非完了例)は無効例として扱った[NC=F(Non-Completer=Failure)アプローチ]。
- 副次評価項目である移植後24週以内に臨床的に意味のあるCMV感染がみられるまでの期間について、投与群別にKaplan-Meier曲線をプロットし、層別因子のCMV感染リスク因子(高リスク/低リスク)で調整したログランク検定を用いて群間差に関する両側p値(名目上のp値)を求めた。
その他の探索的評価項目に関しては、主要評価項目の解析及び副次評価項目の解析と同様の方法を用いて解析した。 - 主要評価項目については、CMV感染のリスク分類(高リスク/低リスク)、幹細胞源(末梢血/骨髄)、HLA適合及びドナータイプ(一致血縁/不一致血縁/一致非血縁/不一致非血縁)、HLA半合致移植(あり/なし)、患者背景(年齢、性別、体重、地域、移植日から無作為割付けまでの日数)、移植前処置レジメン(骨髄破壊/減量強度前処置/骨髄非破壊)及び併用免疫抑制レジメン(シクロスポリン含有及びタクロリムス含有レジメン)別等のサブグループ解析を行うこととした。
- 事後解析については、移植後14週から24週の臨床的に意味のあるCMV感染(晩期CMV感染)の予測因子を算出し、評価資料として承認時に評価された。
解析対象例数:
有効性解析対象例数(FAS):495例(プレバイミス®群325例、プラセボ群170例)
安全性解析対象例数:565例(プレバイミス®群373例、プラセボ群192例)
(うち注射剤の投与を1回以上受けた患者:プレバイミス®群99例、プラセボ群48例)
1. 移植後24週以内に臨床的に意味のあるCMV感染※1がみられた患者の割合(主要評価項目、検証的解析項目)※2
移植後24週以内に臨床的に意味のあるCMV感染がみられた患者(NC=Fアプローチ※2)の割合は、プレバイミス®群37.5%(122/325例)であり、プラセボ群の60.6%(103/170例)と比較して有意に低く、プレバイミス®群の優越性が示されました[群間差:-23.5%、片側p値:p<0.0001、層(高リスク/低リスク)で調整したMantel-Haenszel法(各層の2群の症例数の調和平均で重み付け)]。
■移植後24週以内に臨床的に意味のあるCMV感染がみられた患者の割合(優越性試験、検証的解析結果、主要評価項目、FAS)
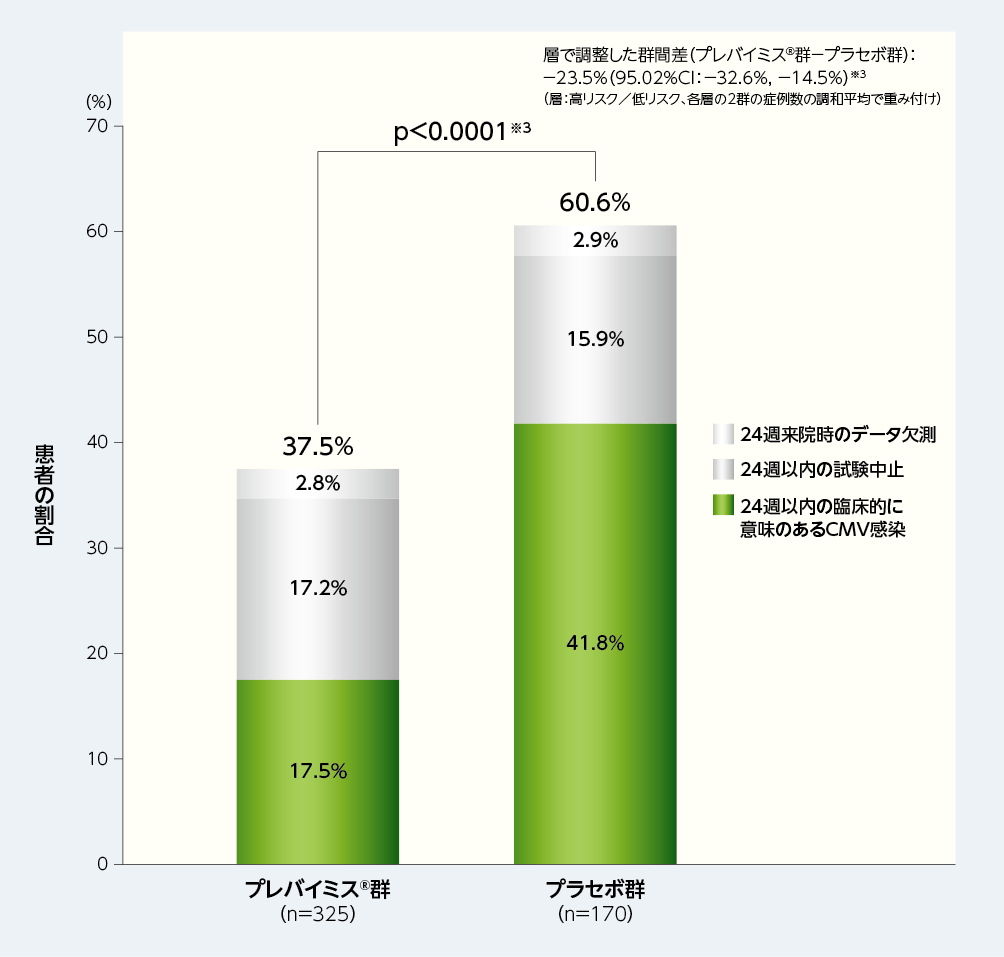
※1:臨床的に意味のあるCMV感染は、「臓器障害を伴うCMV感染症の発症」もしくは「先制治療の開始(中央測定機関でPCR法によりCMV血症が確認された場合及び患者の状態に基づく)」と定義した。
※2:主要評価項目の解析においては、Non-Completer=Failure approach(NC=Fアプローチ)を用いて、24週来院時のデータ欠測例と24週以内の試験中止例は無効例とみなした。
※3:層(高リスク/低リスク)で調整したMantel-Haenszel法(各層の2群の症例数の調和平均で重み付け)を用いて群間差の95.02%CI及びp値を算出し、統計学的有意差の判定には片側p値(0.0249)を用いた。
2. 主要評価項目のサブグループ解析
主要評価項目(移植後24週以内に臨床的に意味のあるCMV感染※1がみられた患者の割合)に関する群間差において、CMV感染のリスク分類や幹細胞源、HLA適合及びドナータイプなどのリスク因子別、移植前処置レジメン及び併用免疫抑制レジメン別にサブグループ解析を行いました。結果は以下の通りでした。
■移植後24週以内に臨床的に意味のあるCMV感染がみられた患者の割合に関する群間差※2
(主要評価項目のサブグループ解析、FAS、DAO※3)
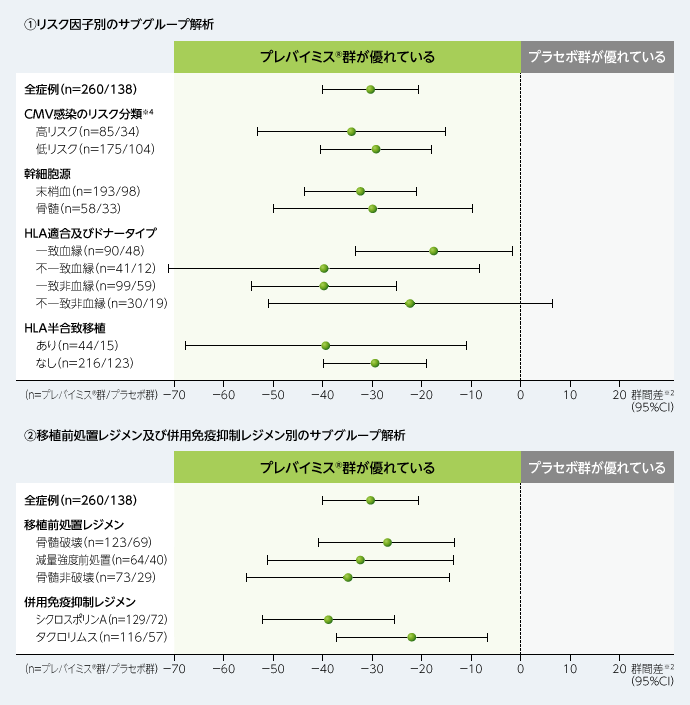
※1:臨床的に意味のあるCMV感染は、「臓器障害を伴うCMV感染症の発症」もしくは「先制治療の開始(中央測定機関でPCR法によりCMV血症が確認された場合及び患者の状態に基づく)」と定義した。
※2:層(高リスク/低リスク)で調整したMantel-Haenszel法(各層の症例数の調和平均で重み付け)を用いて群間差の95%CIを算出した。
※3:Data as Observed:欠測データを補完せず、特定の評価項目に欠測値のある患者は解析から除外した。
※4:高リスク群:無作為割付け時に以下の基準を1つ以上満たす患者
①血縁(同胞)ドナーで、3つのHLA遺伝子座(HLA-A、HLA-B又はHLA-DR)の少なくとも1つに1箇所以上の不一致がある、②HLA半合致ドナー、③非血縁ドナーで、4つのHLA遺伝子座(HLA-A、HLA-B、HLA-C又はHLA-DRB1)の少なくとも1つに1箇所以上の不一致がある、④臍帯血移植、⑤ex vivo T細胞除去移植片の使用、⑥全身性コルチコステロイド(プレドニゾロン換算で1mg/kg/日以上のコルチコステロイド)の使用を必要とするGrade2以上の移植片対宿主病(GVHD)
低リスク群:高リスクの定義に該当しないすべての患者
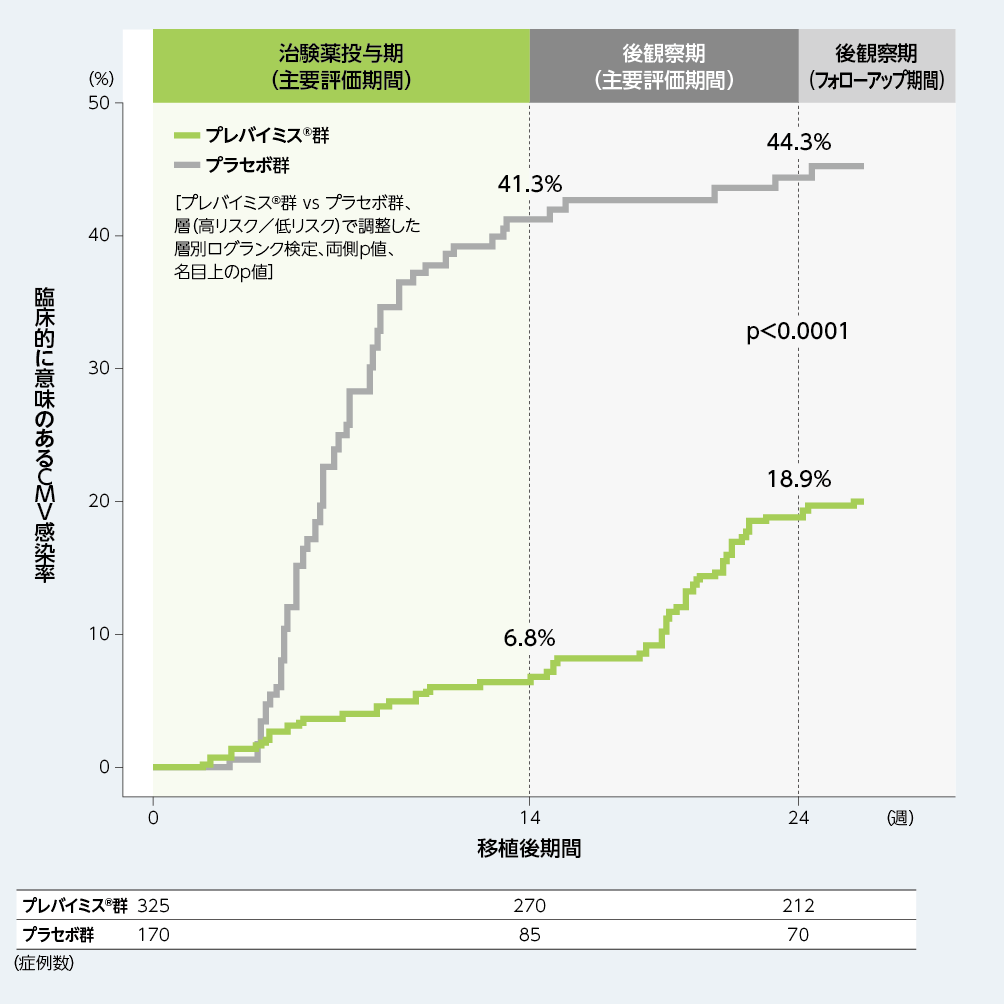
3. 移植後24週以内に臨床的に意味のあるCMV感染がみられるまでの期間(副次評価項目)
移植後24週以内に臨床的に意味のあるCMV感染がみられるまでの期間を、Kaplan-Meier法により評価した結果、プレバイミス®群の移植後24週時点の累積イベント発生率は18.9%であり、プラセボ群の44.3%と比較して両群間で差がみられました[両側p値:p<0.0001、層(高リスク/低リスク)で調整した層別ログランク検定、名目上のp値]。
■移植後24週以内に臨床的に意味のあるCMV感染がみられるまでの期間(副次評価項目、FAS)
4. 移植後14週以内に臨床的に意味のあるCMV感染※1がみられた患者の割合(副次評価項目)※2
移植後14週(治験薬投与期)以内に、臨床的に意味のあるCMV感染がみられた患者(NC=Fアプローチ※2)の割合は、プレバイミス®群が19.1%(62/325例)であり、プラセボ群は50.0%(85/170例)でした[群間差:-31.3%、片側p値:p<0.0001、層(高リスク/低リスク)で調整したMantel-Haenszel法(各層の2群の症例数の調和平均で重み付け)、名目上のp値]。
■移植後14週以内に臨床的に意味のあるCMV感染がみられた患者の割合(副次評価項目、FAS)
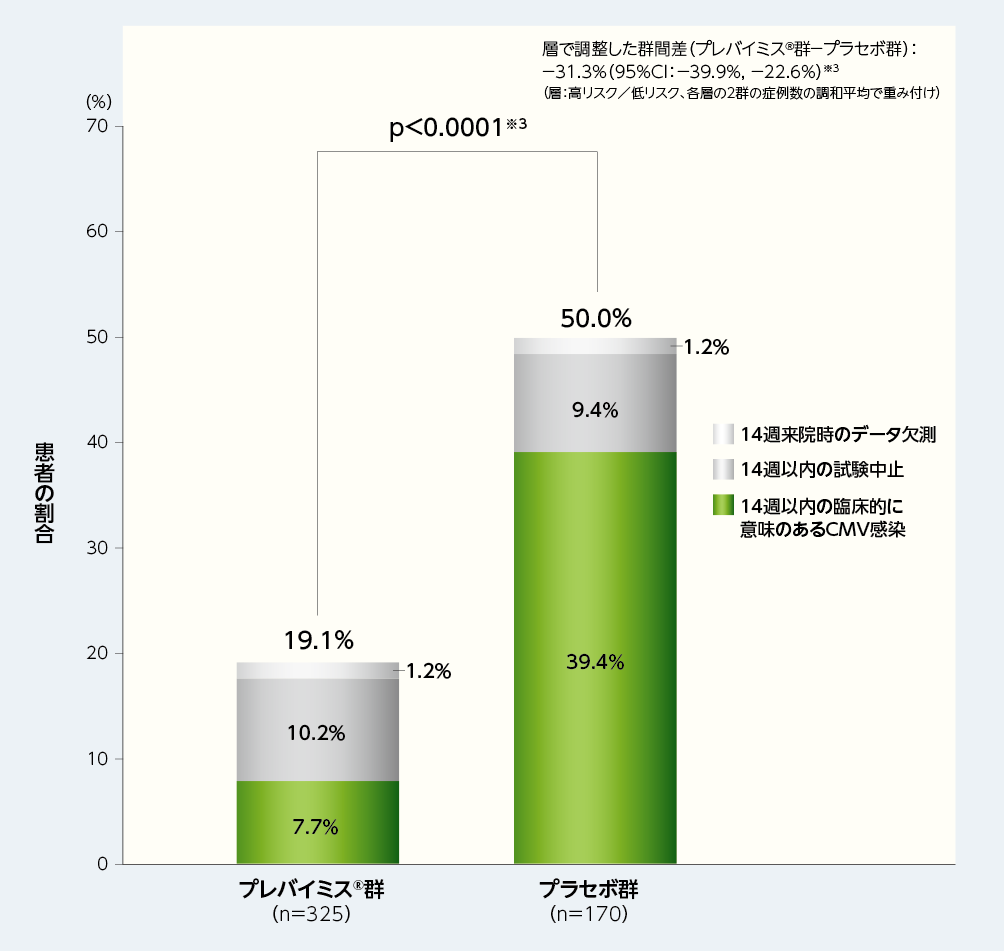
※1:臨床的に意味のあるCMV感染は、「臓器障害を伴うCMV感染症の発症」もしくは「先制治療の開始(中央測定機関でPCR法によりCMV血症が確認された場合及び患者の状態に基づく)」と定義した。
※2:副次評価項目の解析においては、Non-Completer=Failure approach(NC=Fアプローチ)を用いて、14週来院時のデータ欠測例と14週以内の試験中止例は無効例とみなした。
※3:層(高リスク/低リスク)で調整したMantel-Haenszel法(各層の2群の症例数の調和平均で重み付け)を用いて群間差の95%CI及びp値を算出した。p値は、片側p値(名目上のp値)とされ、プレバイミス®の投与とその効果の関連性に対する指標に用いられた。
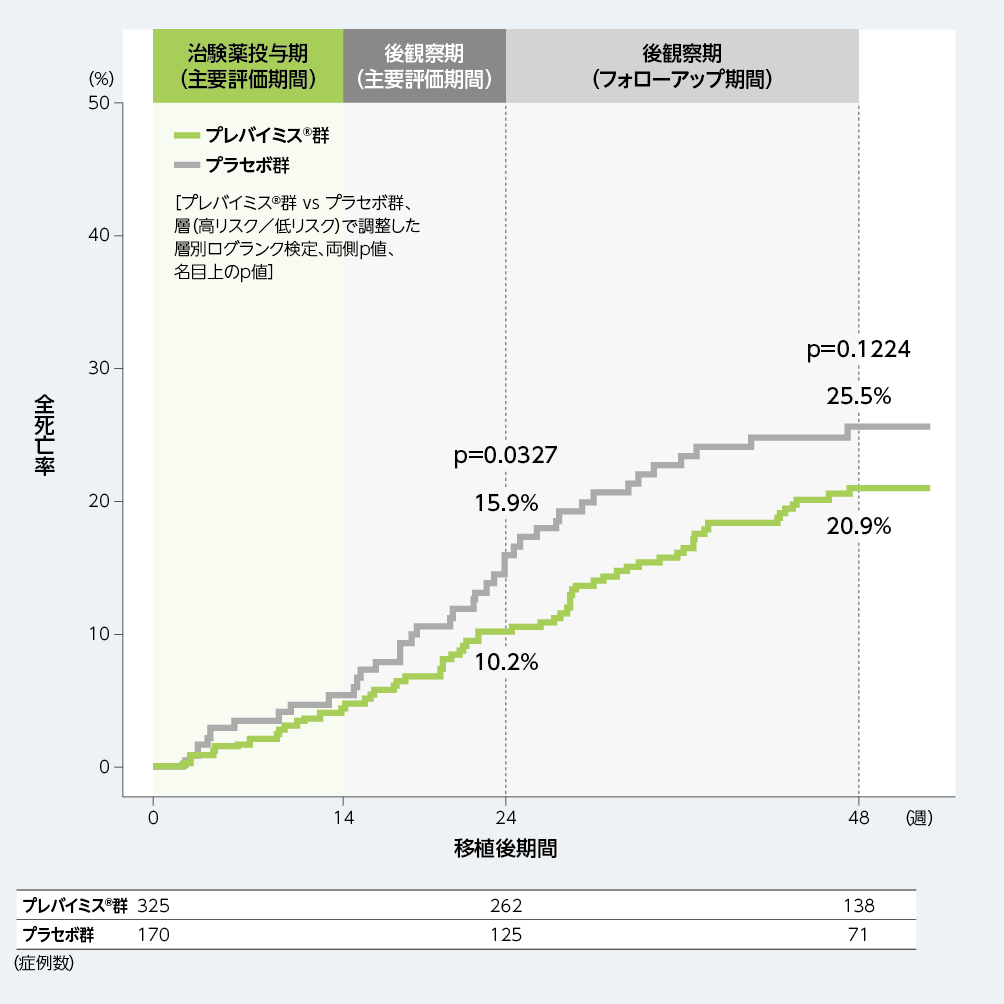
5. 移植後48週以内の全死亡(探索的評価項目)
移植後24週時点のKaplan-Meier法による全死亡の累積発生率は、プレバイミス®群10.2%、プラセボ群15.9%であり、移植後48週時点では、プレバイミス®群20.9%、プラセボ群25.5%でした。
■移植後48週以内の全死亡(探索的評価項目、FAS)
6. 移植後24週以内の生着※1率(探索的評価項目)(参考情報)
移植後24週までのKaplan-Meier法による累積生着率は、プレバイミス®群99.1%、プラセボ群98.7%でした。生着までの期間(中央値)は、プレバイミス®群で19日、プラセボ群で18日でした。
■移植後24週以内の生着率(探索的評価項目、ASaT※2のうち無作為割付け時点で生着が認められなかった患者※3)(参考情報)
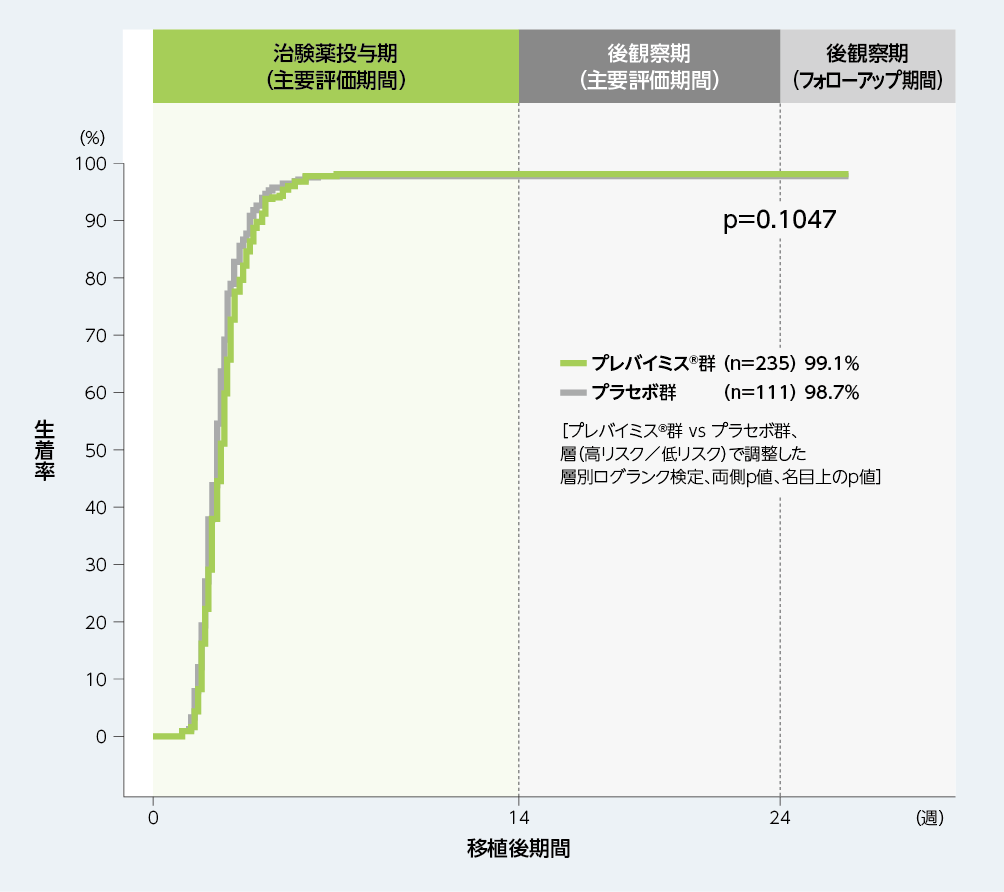
※1:3日連続して好中球絶対数が500/mm3以上の場合と定義した。
※2:All Subjects as Treated;無作為割付け後に治験薬投与を1回以上受けたすべての患者(実際に投与された治験薬に対応する投与群)
※3:無作為割付け時点で生着が認められなかった患者を対象に生着率及び生着までの期間を検討した。
7. 移植後14週から24週の臨床的に意味のあるCMV感染がみられた患者(事後解析)
承認時の評価資料として、移植後14週から24週の臨床的に意味のあるCMV感染(晩期CMV感染)の予測因子を算出する目的で事後解析を実施しました。
移植後14週(投与期間終了時)から24週までの期間に、プレバイミス®群の患者で臨床的に意味のある晩期CMV感染の予測因子として、ベースラインにおける高リスク層(ドナーとの血縁関係、HLAの一致度、幹細胞源及びT細胞除去移植片の使用の有無に基づく)、並びに無作為割付け後の移植片対宿主病(GVHD)の発症及びステロイドの併用投与が関連する可能性が示唆されました。
■移植後14週から24週の臨床的に意味のあるCMV感染がみられた患者(晩期CMV感染)(予後因子別、FAS、DAO※1)

※1:Data as Observed:欠測データを補完せず、特定の評価項目に欠測値のある患者は解析から除外した。
※2:高リスク群:無作為割付け時に以下の基準を1つ以上満たす患者
a. ヒト白血球抗原(HLA)血縁(同胞)ドナーで、3つのHLA遺伝子座(HLA-A、HLA-B又はHLA-DR)の少なくとも1つに1箇所以上の不一致がある
b. HLA半合致ドナー
c. 非血縁ドナーで、4つのHLA遺伝子座(HLA-A、HLA-B、HLA-C又はHLA-DRB1)の少なくとも1つに1箇所以上の不一致がある
d. 臍帯血移植
e. ex vivo T細胞除去移植片の使用
f. 全身性コルチコステロイド(プレドニゾン換算で1mg/kg/日以上のコルチコステロイド)の使用を必要とするGrade 2以上の移植片対宿主病(GVHD)
低リスク群:高リスクの定義に該当しないすべての患者
N:各投与群における患者数 n(%):各サブカテゴリーにおける晩期CMV感染の症例数(%)
8. 安全性
副作用※発現率
移植後24週目までの副作用は、プレバイミス®群で373例中63例(16.9%)、プラセボ群で192例中23例(12.0%)に認められました。両群で頻度の高かった副作用(発現頻度2%以上)は、プレバイミス®群で悪心27例(7.2%)、下痢9例(2.4%)、プラセボ群で悪心7例(3.6%)でした。重篤な副作用は、プレバイミス®群で汎血球減少症、血小板減少症及び生着遅延が各1例、プラセボ群でボーエン病、精神状態変化及び急性腎障害が各1例でした。投与中止に至った副作用は、プレバイミス®群18例(4.8%)、プラセボ群7例(3.6%)で、主なものはプレバイミス®群で悪心6例、嘔吐3例、腹痛2例、プラセボ群で悪心2例でした。
本試験において、死亡に至った副作用は認められませんでした。
■第Ⅲ相国際共同試験(001試験)における副作用発現率(臨床検査値異常含む)
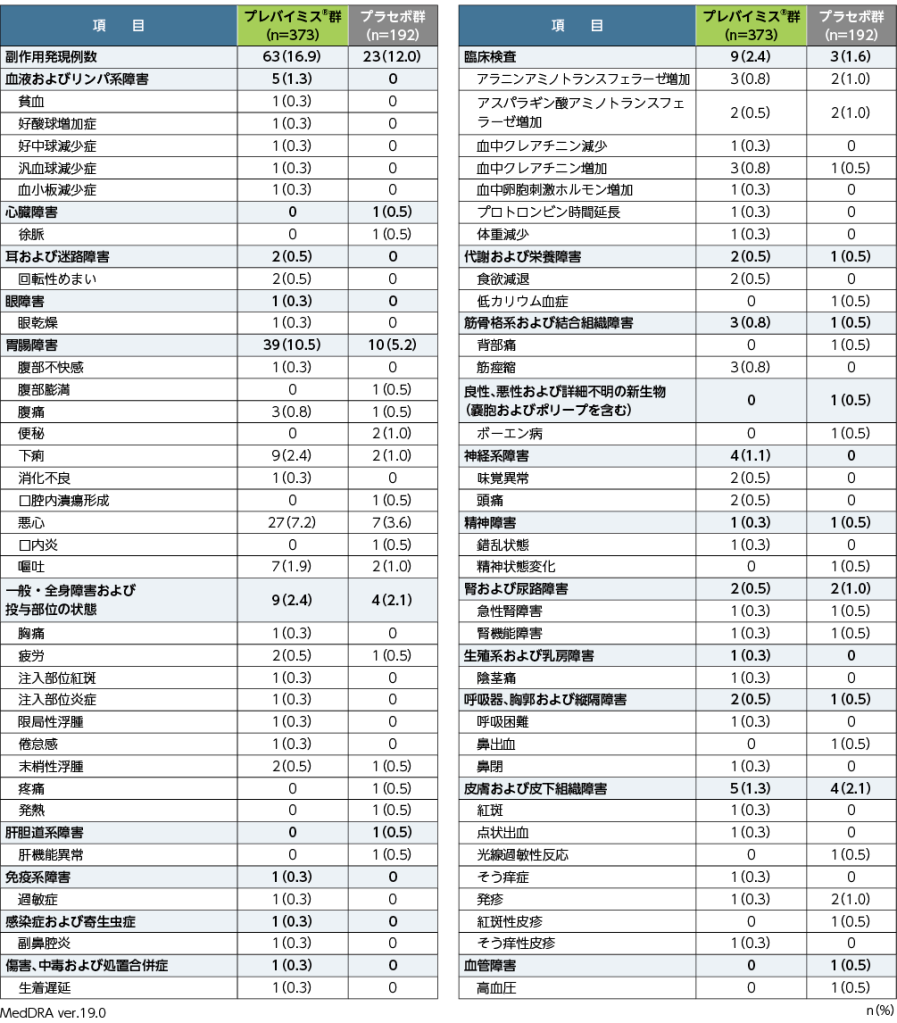
同一患者が2件以上の副作用を発現した場合、同一分類内であれば1件とし、異なる分類であれば、それぞれ1件として集計した。
※:治験担当医師が盲検下で治験薬との因果関係ありと判定した有害事象を副作用と定義した。