製品基本Q&A
製品基本Q&A
プレバイミス®
製品情報
プレバイミス®錠、顆粒分包、点滴静注の電子添文には以下のとおり記載されています。
4. 効能又は効果
下記におけるサイトメガロウイルス感染症の発症抑制
○同種造血幹細胞移植
○臓器移植
5. 効能又は効果に関連する注意
〈臓器移植〉
腎移植以外の臓器移植患者を対象に本剤の有効性及び安全性を評価する臨床試験は実施していない。
レテルモビルはウイルスの複製に必要なCMVのDNAターミナーゼ複合体を阻害します。生化学的な検討及び電子顕微鏡所見から、レテルモビルは一単位長のゲノムの生成に影響し、ウイルス粒子の形成を阻害することが明らかとなりました。
<引用>
電子添文
レテルモビルは、CMV以外の他のウイルスには抗ウイルス作用が認められず、HCMV(ヒトサイトメガロウイルス)に選択的でした(1)(2)。
<検討したウイルス>
他のヘルペスウイルス:水痘・帯状疱疹ウイルス(VZV)、単純ヘルペスウイルス1型及び2型(HSV-1, HSV-2)、ヒトヘルペスウイルス6(HHV-6)、Epstein-Barrウイルス(EBV)
ヘルペスウイルス以外:ヒトアデノウイルス2(HAdV-2)、B型肝炎ウイルス(HBV)、ヒト免疫不全ウイルス1(HIV-1)、A型インフルエンザウイルス、C型肝炎ウイルス(HCV)
<引用>
(1)申請資料概要(2018年03月23日)[HSCT] 2.6.2 p.28-30.
(2)Marschall M et al. Antimicrob Agents Chemother. 2012;56(2):1135-1137.
レテルモビルは体内での代謝を受けにくい薬剤です。
(詳細)
血漿中のレテルモビルの大部分は未変化体として存在し、血漿中に主要代謝物は検出されませんでした。
レテルモビルの全体的な代謝プロファイルから、一部はUGT1A1/1A3を介したグルクロン酸抱合により代謝を受けると考えられます。
<引用>
申請資料概要(2018年03月23日)[HSCT] 2.7.2 p.20
使用方法
プレバイミス®錠、顆粒分包の電子添文には、以下のとおり記載されています。
【プレバイミス®錠240mg】【プレバイミス®顆粒分包20mg/120mg】
通常、成人にはレテルモビルとして 480 mg(240 mg 錠 2 錠又は 120 mg 顆粒 4 包)を 1 日 1 回経口投与する。シクロスポリンと併用投与する場合にはレテルモビルとして 240 mg(240 mg 錠 1 錠又は 120 mg 顆粒 2 包)を 1 日 1 回経口投与する。
通常、小児にはレテルモビルとして以下の用量を 1 日 1 回経口投与する。

7. 用法及び用量に関連する注意
〈効能共通〉
7.1 経口剤(錠剤及び顆粒剤)と注射剤は医師の判断で切り替えて使用することができる。なお、体重30 kg 未満の小児では、切り替える際に用量の調節が必要となる場合がある。
7.2 サイトメガロウイルス血症又はサイトメガロウイルス感染症が確認された場合には、本剤の投与を中止し、サイトメガロウイルスに対する治療等、適切な対応を行うこと。[17.1.1-17.1.5参照]
〈同種造血幹細胞移植〉
7.3 同種造血幹細胞移植の移植当日から移植後28日目までを目安として投与を開始すること。投与期間は、患者のサイトメガロウイルス感染症の発症リスクを考慮しながら、移植後200日目までを目安とすること。[17.1.1-17.1.3参照]
〈臓器移植〉
7.4 移植後早期より投与を開始し、投与期間は、患者のサイトメガロウイルス感染症の発症リスクを考慮しながら、移植後200日目までを目安とすること。ただし、レテルモビルは主に肝を介して消失するため、移植後に肝機能が安定しない場合、血漿中濃度が上昇するおそれがあることから、投与可否を慎重に判断すること。 [9.3.1、16.6.2、17.1.4、17.1.5参照]
プレバイミス®点滴静注の電子添文には、以下のとおり記載されています。
【プレバイミス®点滴静注240mg】
6. 用法及び用量
通常、成人にはレテルモビルとして 480 mg を 1 日 1 回、約 60 分かけて点滴静注する。シクロスポリンと併用投与する場合にはレテルモビルとして 240 mg を 1 日 1 回、約 60 分かけて点滴静注する。
通常、小児にはレテルモビルとして以下の用量を 1 日 1 回、約 60 分かけて点滴静注する。
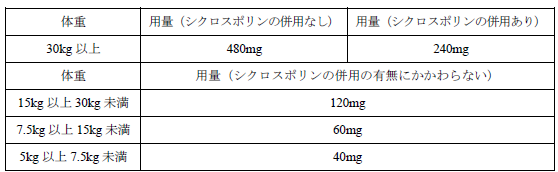
7. 用法及び用量に関連する注意
〈効能共通〉
7.1 経口剤(錠剤及び顆粒剤)と注射剤は医師の判断で切り替えて使用することができる。なお、体重 30 kg 未満の小児では、切り替える際に用量の調節が必要となる場合がある。ただし、臨床試験において注射剤の長期投与の経験はなく、注射剤の添加剤ヒドロキシプロピル-β-シクロデキストリンは腎機能障害のある患者で蓄積し、腎機能の悪化等を引き起こすおそれがあることから、注射剤の投与は最小限の期間とし、経口投与可能な患者には、経口投与を選択すること。[9.2.1 参照]
7.2 サイトメガロウイルス血症又はサイトメガロウイルス感染症が確認された場合には、本剤の投与を中止し、サイトメガロウイルスに対する治療等、適切な対応を行うこと。[17.1.1-17.1.5参照]
〈同種造血幹細胞移植〉
7.3 同種造血幹細胞移植の移植当日から移植後 28 日目までを目安として投与を開始すること。投与期間は、患者のサイトメガロウイルス感染症の発症リスクを考慮しながら、移植後 200 日目までを目安とすること。[17.1.1-17.1.3 参照]
〈臓器移植〉
7.4 移植後早期より投与を開始し、投与期間は、患者のサイトメガロウイルス感染症の発症リスクを考慮しながら、移植後 200日目までを目安とすること。ただし、レテルモビルは主に肝を介して消失するため、移植後に肝機能が安定しない場合、血漿中濃度が上昇するおそれがあることから、投与可否を慎重に判断すること。[9.3.1、16.6.2、17.1.4、17.1.5 参照]
電子添文「7.用法及び用量に関連する注意」に記載されている「同種造血幹細胞移植の移植当日から移植後28日までを目安として投与開始」は、第Ⅲ相臨床試験[001試験(HSCT 100日投与)](*)で規定したプレバイミス®の投与期間に基づき設定しました。
<なぜ001試験(HSCT 100日投与)でそのように設定したのか?>
本剤が予防投与を目的としていることから、CMV感染が起こるよりも前の時期、早ければ移植当日、遅くとも移植後28日目までに生着の有無にかかわらず投与を開始することが望ましいと考えました(1)。
(*)001試験 (HSCT 100日投与): 成人HSCT患者を対象に臨床的に意味のあるCMV 感染の予防を目的としてプレバイミスを移植後100日目まで投与した第Ⅲ相試験(二重盲検, 無作為化, プラセボ対照)
<引用>
(1)申請資料概要(2018年03月23日)[HSCT] 1.8.3 p.11
電子添文上、投与期間は「患者のサイトメガロウイルス感染症の発症リスクを考慮しながら」と規定されています。
よって、200日目までを目安に投与することの必要性については発症リスクに応じて患者ごとに主治医の判断をお願いします。
参考までに、臨床試験において同種造血幹細胞移植後100日目まではCMV抗体陽性レシピエントが投与対象とされ、移植後100日以降200日目までは、他にもCMV感染症のリスク因子を有する、よりハイリスクな患者が投与対象とされました。
投与期間設定の参考となるCMV感染症のリスク因子については、本剤の第Ⅲ相臨床試験[040試験(HSCT 200日投与)](*)の組み入れ基準(1)、日本造血・免疫細胞療法学会のガイドライン(2)、ならびに実地臨床研究の結果(3)など、最新の知見も考慮の上、主治医の判断をお願いします。
(*)040試験 (HSCT 200日投与): 成人HSCT患者へのプレバイミス予防投与を移植後100日から200日に延長した第Ⅲ相試験 (二重盲検, 無作為化, プラセボ対照)
<引用>
(1)電子添文
(2)造血細胞移植ガイドライン サイトメガロウイルス感染症(第5版)2022年6月, 発行:日本造血・免疫細胞療法学会, 2022
(3)Mori Y et al. Int J Hematol. 2022;116(2):258-265.
患者のサイトメガロウイルス感染症の発症リスクを考慮して投与期間を設定してください。
腎移植患者を対象とした第Ⅲ相臨床試験(1)(2)の結果から、200日投与での有効性と安全性が評価されたことに基づき、投与期間の目安を200日としています。
具体的な投与期間は、臓器移植患者を対象とした第Ⅲ相臨床試験(1)(2)、臓器移植関連CMV感染症診療ガイドライン(3)等を参考に、移植臓器や患者背景に応じて個別にご判断ください。
<引用>
(1)Limaye AP et al. JAMA. 2023;330(1):33-42.
(2)Ishida H et al. Clin Exp Nephrol. 2024;28(8):822-831.
(3)臓器移植関連CMV感染症 診療ガイドライン. 発行:日本移植学会, 2022:107.
本剤の電子添文には、以下のとおり記載されています。
2. 禁忌(次の患者には投与しないこと)
2.1 本剤の成分に対し過敏症の既往歴のある患者
2.2 次の薬剤を投与中の患者:ピモジド、エルゴタミン酒石酸塩・無水カフェイン・イソプロピルアンチピリン、ジヒドロエルゴタミン、メチルエルゴメトリン、エルゴメトリン[10.1 参照]
【適応外】
CMV感染の治療(先制治療)/感染症の治療(標的治療)に対する使用は適応外です。
本剤のご使用にあたっては電子添文をご確認ください。
CMV感染の治療(先制治療)/感染症の治療(標的治療)に関して有効性、安全性を十分に検証したデータはありません。
【適応外】
同種造血幹細胞移植、臓器移植以外の患者への使用は適応外であり、有効性及び安全性データがありません。
本剤のご使用にあたっては電子添文をご確認ください。
本剤の電子添文の「9.特定の背景を有する患者に関する注意」には、以下のとおり記載されています。
9.4 生殖能を有する者
妊娠可能な女性に対しては、本剤が胎児に悪影響を及ぼす可能性があることを十分に説明し、本剤投与中及び本剤投与終了後一定期間は適切な避妊を行うよう指導すること。[9.5 参照]
9.5 妊婦
【プレバイミス®錠240mg】【プレバイミス®顆粒分包20mg/120mg】
妊婦又は妊娠している可能性のある女性には、本剤投与の有益性が危険性を上回ると判断される場合にのみ投与すること。妊娠中に本剤を投与するか、本剤投与中の患者が妊娠した場合は、本剤投与による催奇形性等が生じる可能性があることについて、患者に十分説明すること。
妊娠ラット及びウサギの器官形成期に投与したとき、成人同種造血幹細胞移植患者の臨床曝露量(シクロスポリン併用下での240mg経口投与)のそれぞれ18倍及び2.8倍の母動物毒性を示す用量で骨格奇形、胎児体重の減少等が認められた。妊娠ラットに着床から分娩後まで投与した試験では、臨床曝露量の3.7倍まで胚・胎児毒性は認められなかった。[9.4 参照]
【プレバイミス®点滴静注240mg】
妊婦又は妊娠している可能性のある女性には、本剤投与の有益性が危険性を上回ると判断される場合にのみ投与すること。妊娠中に本剤を投与するか、本剤投与中の患者が妊娠した場合は、本剤投与による催奇形性等が生じる可能性があることについて、患者に十分説明すること。
妊娠ラット及びウサギの器官形成期に投与したとき、成人同種造血幹細胞移植患者の臨床曝露量(480mg静脈内投与)のそれぞれ11倍及び1.7倍の母動物毒性を示す用量で骨格奇形、胎児体重の減少等が認められた。妊娠ラットに着床から分娩後まで投与した試験では、臨床曝露量の2.2倍まで胚・胎児毒性は認められなかった。[9.4参照]
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。動物試験(ラット)で乳汁移行が認められている(1)。
<引用>
(1)申請資料概要(2018年03月23日)[HSCT] 2.6.4.6
電子添文上では用量調節するタイミングについて明確な規定はありません。
ご参考までに、本剤の投与開始後にシクロスポリンの使用を新たに開始又は中止する場合、米国及びEUの添付文書(1)(2)では次回の投与から本剤の用量を変更することが規定されています。
ただし、シクロスポリンの血中濃度の上昇によりシクロスポリンを一時的に休薬する場合は、プレバイミス®240mgの用量は変更しないこととされています。
<引用>
(1)米国添付文書
(2)EU添付文書
レテルモビルを成人健康被験者に720mg/日から1,440mg/日を最長14日間投与した際に認められた副作用は、推奨用量である480mg/日を投与した場合と類似していました。過量投与が生じた際は、患者に副作用の徴候がないか観察し、必要に応じ適切な対症療法を実施してください。
<引用>
インタビューフォーム Ⅷ.安全性(使用上の注意等)に関する項目 10.過量投与
経口剤(錠剤、顆粒剤)と注射剤は医師の判断で切り替えて使用することができます。
ただし、臨床試験において注射剤の長期投与の経験はなく、注射剤の添加剤ヒドロキシプロピル-β-シクロデキストリンは腎機能障害のある患者で蓄積し、腎機能の悪化等を引き起こすおそれがあることから、注射剤の投与は最小限の期間とし、経口投与可能な患者には、経口投与を選択するよう、注射剤の電子添文に記載されています。
なお、臨床試験(001試験、002試験、040試験、042試験、030試験)では原則として、治験薬は経口剤を投与することとし、経口剤の投与が難しいと思われる患者(*)にのみ注射剤を用いていました(1)。
(*)嘔吐や下痢、吸収不能など、患者が嚥下不能又は錠剤の吸収を妨げる可能性のある状態
<引用>
(1)インタビューフォーム V. 治療に関する項目
成人同種造血幹細胞移植患者を対象とした第Ⅲ相臨床試験[001試験(HSCT 100日投与)](*1)の結果より、製剤(錠剤、注射剤)の違い及び切替えの有無にかかわらず、投与期間が十分であれば一貫した有効性が得られることが示唆されています(1)。
なお、顆粒剤については錠剤と生物学的同等性を示すことが確認されています(2)。
(*1)001試験 (HSCT 100日投与): 成人HSCT患者を対象に臨床的に意味のあるCMV 感染の予防を目的としてプレバイミスを移植後100日目まで投与した第Ⅲ相試験(二重盲検, 無作為化, プラセボ対照)
<引用>
(1)申請資料概要(2018年03月23日)[HSCT] 2.5 p.60.
(2)申請資料概要[小児] 2.7.1.2.1 レテルモビルの顆粒剤と錠剤の相対的バイオアベイラビリティ試験
プレバイミス® 錠剤、顆粒剤とも食事に関係なく投与することができます。
<引用>
(1)申請資料概要(2018年03月23日)[HSCT] 2.7.6 個々の試験のまとめ p.54
(2)申請資料概要[小児] 2.7.1.4 結論
【適応外】
本剤を粉砕して投与することは、承認外の用法となります。
錠剤、顆粒剤とも粉砕して投与した際の薬物動態、有効性、安全性は検討していませんので、おすすめしていません(1)。
粉砕後の安定性データはありません。
<引用>
(1)インタビューフォーム ⅩⅢ.備考 1. 調剤・服薬支援に際して臨床判断を行うにあたっての参考情報
【適応外】
本剤を分割して投与することは、承認外の用法となります。
錠剤分割後の安定性について検討していませんので、おすすめしていません。
錠剤に割線はありません。
【プレバイミス®錠240mg】
プレバイミス錠を分包、又は他剤と一包化した際の安定性について検討していませんので、おすすめしていません。
電子添文「14. 適用上の注意」に記載されているとおり、 PTP シートのまま保存し、服用直前に PTP シートから取り出すよう指導してください。
【プレバイミス®顆粒分包20mg/120mg】
プレバイミス顆粒を開封後、再分包、あるいは他剤と一包化した際の安定性について検討していませんので、おすすめしていません。
電子添文「14. 適用上の注意」に記載されているとおり、スティックパックのまま保存し、服用直前にスティックパックから取り出すよう指導してください。
【適応外】
錠剤、顆粒剤とも簡易懸濁して投与することは、承認外の用法となります。
簡易懸濁にて投与した際の薬物動態、有効性、安全性は検討していませんので、おすすめしていません。
簡易懸濁後の安定性データはありません。
安全性
主な副作用は、血液及びリンパ系障害として白血球減少症 (1%以上5%未満) 、好中球減少症 (1%未満)、胃腸障害として悪心、下痢(1%以上5%未満)、嘔吐(1%以上5%未満)、免疫系障害として過敏症 (1%未満)、臨床検査として白血球数減少 (1%未満)です(1)。
<引用>
(1)インタビューフォーム Ⅰ.概要に関する項目 2.製品の治療学的特性
その他
プレバイミス®錠剤、プレバイミス®顆粒剤の電子添文には、以下のとおり記載されています。
14. 適用上の注意
14.1 薬剤交付時の注意
〈錠剤〉
14.1.1 PTP シートのまま保存し、服用直前に PTP シートから取り出すよう指導すること。
14.1.2 PTP 包装の薬剤は PTP シートから取り出して服用するよう指導すること。PTP シートの誤飲により、硬い鋭角部が食道粘膜へ刺入し、更には穿孔をおこして縦隔洞炎等の重篤な合併症を併発することがある。
〈顆粒剤〉
14.1.3 スティックパックのまま保存し、服用直前にスティックパックから取り出すよう指導すること。
14.1.4 本剤は軟らかい食品に混ぜて経口投与することが望ましい。また、経口投与が困難な場合は経鼻又は胃瘻チューブを介して投与できる。患者又は保護者等に対し、患者用説明文書を参照するよう指導すること。
・経鼻又は胃瘻チューブを介して投与する場合、本剤を室温の液体注)が入った容器に入れて10分程度静置する。顆粒が崩壊した後にシリンジで混合して、全量をシリンジ、及び経鼻又は胃瘻チューブを用いて投与する。
・その後、容器やシリンジを室温の液体注)ですすぎ、すすいだ全量をシリンジ、及び経鼻又は胃瘻チューブを用いて投与する。
・最後に、経鼻又は胃瘻チューブに水を流して投与する。
注)胃瘻チューブを介して投与する場合、チューブ内に本剤が残存する可能性があるため、水の使用は推奨されない。
患者さんが通常食べられるような軟らかい食品であれば、本剤と混ぜて投与することが可能です。
ただし、熱い食品は用いないでください。
<経鼻チューブで投与する場合 (1)(2)>
牛乳、リンゴジュース、調製ミルク、又は水での投与をお願いします。
<胃瘻チューブで投与する場合 (1)(2)>
牛乳、リンゴジュース、調製ミルクでの投与をお願いします。
胃瘻チューブを使用する場合はチューブ内に薬剤が残留する可能性があるため、水の使用は推奨されません。
<引用>
(1)患者用説明文書
(2)インタビューフォーム Ⅷ. 安全性(使用上の注意等)に関する項目 11. 適用上の注意
プレバイミス®点滴静注240mgの電子添文には、以下のとおり記載されています。
14. 適用上の注意
14.1 薬剤調製時の注意
14.1.1 希釈前に、変色や不溶性異物がないか、各バイアルを確認すること。本剤は無色澄明の溶液である。また、製品由来の少量の半透明又は白色の微粒子を含むことがある。バイアル内の溶液に変色や濁り、又は異物(少量の半透明又は白色の微粒子以外)が認められた場合は使用しないこと。バイアルを振盪しないこと。
14.1.2 バイアルから溶液を採取して日局生理食塩液又は日局 5%ブドウ糖注射液が入った点滴バッグに添加し、振盪せず静かに混和すること。
60~480 mg を投与する場合、バイアルからの採取液量、点滴バッグの液量は以下を参照し、点滴バッグの総液量を投与すること。

40 mg を投与する場合、1 バイアル(レテルモビル濃度 20 mg/mL)から 5 mL を採取し、45 mL の日局生理食塩液又は日局 5%ブドウ糖注射液が入った点滴バッグに添加し、振盪せず静かに混和すること。当該希釈液を 20 mL 投与すること。
14.1.3 本剤のバイアルは 1 回使い切りである。残液は使用しないこと。
14.1.4 混和後、本剤の希釈液は無色~黄色澄明の溶液となる。投与前の希釈液に変色や不溶性異物がないか目視により確認すること。変色や濁り、又は異物(少量の半透明又は白色の微粒子以外)が認められる場合には、希釈液を廃棄すること。
14.1.5 希釈液は、室温保存(2~30℃)では 24 時間以内に、冷蔵保存(2~8℃)した場合は 48 時間以内に使用すること。なお、これらの時間には点滴終了までの時間が含まれる。
14.2 配合変化
本剤は他剤と配合したとき、濁りや不溶性異物が生じることがある。配合適性についてはデータが限られているが、次の薬
剤は配合禁忌であり、同一の輸液ラインを通して同時に注入しないこと。
主な配合禁忌薬剤:アミオダロン塩酸塩、アムホテリシン B リポソーム、アズトレオナム、セフェピム塩酸塩、シプロフロキサシン、シクロスポリン、ジルチアゼム塩酸塩、フィルグラスチム(遺伝子組換え)、ゲンタマイシン硫酸塩、レボフロキサシン、リネゾリド、ミダゾラム、オンダンセトロン塩酸塩、パロノセトロン塩酸塩
14.3 薬剤投与時の注意
14.3.1 必ず 0.2 μm インラインフィルター(ポリエーテルスルホン、ポリスルホン又は正荷電ナイロン製)を使用して投与すること。
14.3.2 本剤はポリウレタンを含有する輸液チューブで投与しないこと。
240mgを125mLに希釈した場合、480mgを250mLに希釈した場合と同一の濃度であることから、濃度による問題はありません。
しかしながら、第Ⅲ相試験において、用量(480mg又は240mg)を問わず250mLに希釈した際の有効性及び安全性を評価していることから、電子添文ではいずれも250mLへの希釈と規定しています。
ポリウレタンを含有する輸液チューブとの適合性を検討したところ、レテルモビルの定量値が低下することが確認されたため、ポリウレタンを含有する輸液チューブで投与しないこととしました。
電子添文上特に規定はありませんが、続けて他の薬剤を投与する場合には、生理食塩液でフラッシュして使用することをおすすめします。
【プレバイミス®錠240mg】【プレバイミス®顆粒分包20mg/120mg】
貯法は、室温保存です。
【プレバイミス®点滴静注240mg】
貯法は、室温保存です。
外箱開封後は遮光して保存してください。
<引用>
電子添文