製品基本Q&A
製品基本Q&A
ジーンプラバ®
製品情報
本剤の添付文書には以下のとおり記載されています。
4.効能又は効果
クロストリジウム・ディフィシル感染症の再発抑制
5.効能又は効果に関連する注意
臨床成績の項の内容を理解した上で、クロストリジウム・ディフィシル感染症の既往がある等、再発リスクが高いと判断した患者を対象とすること。[17.1.1参照]
<引用>
添付文書
ベズロトクスマブは、クロストリジウム・ディフィシルが産生するトキシンBに対する中和抗体です。
ベズロトクスマブが、トキシンBに特異的に結合することによって、トキシンBと宿主細胞との結合を阻害し、トキシンBによる腸壁の炎症及び損傷を抑制し、CDIの再発を抑制します。
<CDIにおけるトキシンBの役割について>
クロストリジウム・ディフィシルの病原因子であるトキシンA及びトキシンBが、宿主細胞の受容体に結合し、細胞内に取り込まれることで、腸管上皮のバリアが破壊されます。
トキシンの細胞傷害作用及び炎症性メディエーターの放出により、腸管上皮の損傷及び炎症性反応を惹起してCDIを引き起こします。
近年では、トキシンBが臨床上、病原性に重要な役割を担っていることが示唆されています。
<引用>
インタビューフォーム
ベズロトクスマブはトキシンB に選択的であり、トキシンAには作用しないと考えられています。
廃棄量を可能な限り少なくする目的のため625mgを設定しました。
なお、第Ⅲ相試験に組み入れられた日本人のデータでは、約9割程度は625mgでカバーされています。
In vitro試験において、ベズロトクスマブは、国内外で得られたクロストリジウム・ディフィシルの臨床分離株(リボタイプ001/072、002、003、012、014、017、018、023、027、052、053、063、077、078、081、087、106、198及び369)に由来するトキシンBによる細胞傷害作用を阻害しました。
<引用>
添付文書
インタビューフォーム
抗菌活性はありません。
使用方法
本剤の添付文書には以下のとおり記載されています。
6.用法及び用量
通常、成人にはベズロトクスマブ(遺伝子組換え)として10mg/kgを60分かけて単回点滴静注する。
7.用法及び用量に関連する注意
本剤の使用に際しては、次の点に注意すること。
・クロストリジウム・ディフィシル感染症に対する治療は、別途適切に行うこと。
・本剤は、クロストリジウム・ディフィシル感染症に対する治療施行中に投与すること。
・本剤を複数回投与した場合のベネフィット・リスクは不明である。
<引用>
添付文書
本剤の成分に対し過敏症の既往歴のある患者です。
<引用>
添付文書
高齢者における本剤の用量調節は不要です。
母集団薬物動態解析により検討した結果、年齢は本剤の曝露量に対して臨床的に意味のある影響を及ぼしませんでした。
<引用>
インタビューフォーム Ⅶ.薬物動態に関する項目
本剤の添付文書には、以下のとおり記載されています。
9. 特定の背景を有する患者に関する注意
9.5 妊婦
妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断される場合にのみ投与すること。
ヒトIgGは母体から胎児へ移行することが知られている。なお、生殖発生毒性試験は実施していない。
9.6 授乳婦
治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討すること。ヒトIgGは乳汁中に移行することが知られている。本剤がヒト乳汁中へ移行するかは不明である。
18歳未満の患者を対象とした臨床試験は実施していません。
<引用>
添付文書
もともと120分間の点滴静注にて開発を進めていましたが、臨床での利便性向上等を考慮し、60分間での点滴静注による忍容性や薬物動態を検討しました(第Ⅰ相試験:005試験[60分間]、020試験[120分間])。
その結果、60分投与にて、120分とほぼ同程度の曝露量(AUC0-∞)や良好な忍容性が確認できたことから、第Ⅲ相試験では60分間かけての点滴静注にて、有効性・安全性を確認し、用法・用量が設定されました。
ジーンプラバ®の添付文書上、再投与に関連した規定はありません。
本剤投与後に再発した患者に対する再投与の臨床試験は行われておりません。 そのため、再投与による有効性・安全性は不明です。
CDI発症後の期間別にジーンプラバ®の有効性・安全性を検討した報告はなく、わかりません。
同一の点滴ラインで他の薬剤と同時投与はしないでください(1)。
国内治験時でも、同一の点滴ラインで同時併用することを禁止していました。
同じラインで投与する場合は、生理食塩液などでフラッシュした後に、次の薬剤を投与するよう規定していました。
<引用>
(1)添付文書
CDI標準抗菌薬の投与期間中にジーンプラバ®を投与してください。
投与のタイミングによる効果の差はほとんど無いと考えられます。
【適応外】
ボーラス投与(ワンショット静注)は承認されている使用法ではありません。
本剤のご使用にあたっては製品添付文書をご覧いただきますようお願いいたします。
ジーンプラバ®をワンショット静注することはできません。
用法・用量には、「60分かけて単回点滴静注する。」と記載されています。
CDIに対する治療薬と併用することが、添付文書 「7. 用法及び用量に関連する注意」で規定されています。
また、ヒトに単独で投与して、有効性・安全性を検討した報告はありません。
ジーンプラバ®はCDIの再発抑制を適応とする薬剤であるため、どのくらいの期間効果が持続するかを明確に示すことは困難ですが、第Ⅲ相試験では、プラセボを対照に、投与後12週間の再発抑制効果が認められました。
また、ジーンプラバ®投与によって、CDI 再発が遅延するのか、又はCDI 再発が抑制されるのかを評価するため、投与後12ヵ月目まで延長したところ、ジーンプラバ®投与群(n=99)にて、CDI再発は認められませんでした。
<引用>
申請資料概要 2.7.3 臨床的有効性 2.7.3.5 効果の持続、耐薬性
安全性
海外第Ⅲ相試験(001試験)において、ジーンプラバ®投与後4週間に、安全性の評価となった390例中32例(8.2%)に副作用が認められました。主な副作用は浮動性めまい4例(1.0%)、悪心3例(0.8%)及び頭痛3例(0.8%)でした。
国際共同第Ⅲ相試験(002試験)において、ジーンプラバ®投与後4週間に、安全性の評価となった396例(日本人29例を含む)中27例(6.8%)に副作用が認められました。主な副作用は悪心5例(1.3%)、疲労4例(1.0%)及び頭痛3例(0.8%)でした。
<引用>
添付文書
その他
本剤の添付文書には、以下のとおり記載されています。
14.適用上の注意
14.1 薬剤調製時の注意
14.1.1 バイアルは冷所(2~8℃)から取り出したら速やかに調製すること。保存を必要とする場 合には、バイアルは常温、遮光条件下で24時間以内に調製すること。[20.参照]
14.1.2 調製前に変色、異物がないことを確認する。本剤は、無色~うすい黄色で澄明~うすい乳白色の液である。溶液に変色や異物があった場合は使用しないこと。
14.1.3 バイアルは振盪しないこと。
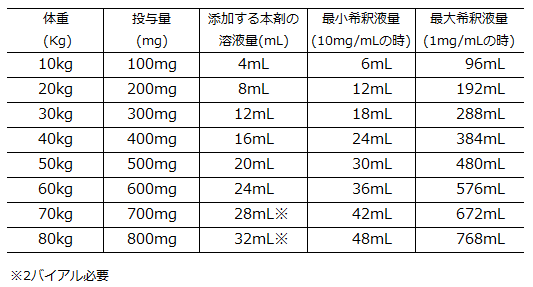
14.1.4 患者の体重に基づいて必要量を分取し、生理食塩液又は5%ブドウ糖注射液を含む点滴バッグに加えて希釈し、最終濃度を1~10mg/mLとする。
14.1.5 希釈後は静かに転倒混和する。
14.1.6 使用後のバイアルと残液は廃棄すること。
14.2 希釈後の保存に関する注意
14.2.1 本剤は保存剤を含まないため、調製後は速やかに使用すること。保存を必要とする場合には、調製開始後、常温では16時間以内、冷所(2~8℃)では24時間以内に使用すること。冷所保存した場合は、投与前に常温に戻すこと。保存可能時間には、点滴終了までの時間を含む。
14.2.2 希釈後は冷凍しないこと。
14.3 薬剤投与時の注意
14.3.1無菌処理及び発熱物質に対する処理が行われたタンパク質低吸着性のインラインフィルター(0.2~5μm)を使用すること。
14.3.2 他の薬剤と同じラインで同時に本剤を投与しないこと。
<引用>
添付文書
貯法は、2~8℃です。
外箱開封後は、遮光して保存してください。
<引用>
添付文書
添付文書上、投与前に必要な検査に関する記載はありません。
医師が再発リスクが高いと判断したCDI患者が投与の対象となります。
なお、保険給付については、各都道府県の審査の先生方の判断が異なる場合もある点、ご注意ください。
主要な海外ガイドライン(1)(2)(3)(4)では、CDIの再発は(発症又は治療終了後から)8週以内に起こるものと定義されています。
ジーンプラバ®の第Ⅲ相臨床試験では、全投与群を通して、CDI再発の大半(約71%)が投与後4週間以内に発生しました(5)。
<引用>
(1)Surawicz CM et al. Am J Gastroenterol. 2013;108(4):478-498. quiz 499
(2)Debast SB et al. Clin Microbiol Infect. 2014;20(Suppl 2):1-26.
(3)Sartelli M et al. World J Emerg Surg. 2015;10:38.
(4)Trubiano JA et al. Intern Med J. 2016;46(4):479-493.
(5)申請資料概要 2.5臨床に関する概括評価 2.5.4.3.1.3 CDI 再発までの期間
表示量としてベズロトクスマブ625mg(容量25mL)を含有しています。
なお、損失を考慮して12.5mg(容量0.5mL)が過量充填されており、総含有量は637.5mgです。
便移植はプロトコール上禁止になっていたため、含まれていません。
<引用>
Wilcox MH et al. N Engl J Med. 2017;376(4):305-317.
(Supplementary MaterialのProtocol p.28-29)
他の薬剤との配合変化を検討したデータはありません。
添付文書においても、他の薬剤と同じラインで同時に本剤を投与しないよう注意喚起しています。
遺伝子組換え技術により、チャイニーズハムスター卵巣細胞を用いて製造されます。
下痢:24時間以内に3回以上の軟便
軟便:ブリストル便形状スケールタイプ5~7
<引用>
申請資料概要 2.7.3 臨床的有効性 2.7.3.1.2.3 有効性評価項目 p.18
申請資料概要 2.5臨床に関する概括評価 p.37
バイアル中に残った残液は、廃棄してください(1)。
ジーンプラバ®の残液に特有の廃棄方法は定められていません。ご施設の基準に沿って廃棄してください。
<引用>
(1) 添付文書
5±3℃の長期保存試験で、36ヵ月の安定性が確認されたため、貯法は2~8℃と設定されました。
一般に、抗体製剤は熱に不安定である可能性が高く、ジーンプラバ®も加速試験(25℃)、苛酷試験(40℃)にて変化が認められています。
生理食塩液又は5%ブドウ糖注射液を用いて、希釈してください。
第Ⅲ相試験において、リスク因子を1つ以上有していた患者は、リスク因子がなかった患者と比べて、CDI再発率が高値でした。(プラセボ群 29.8% vs 16.8%)
<引用>
添付文書