臨床成績
臨床成績
第Ⅲ相二重盲検比較試験
(MODIFY Ⅰ;001 試験(海外データ)/MODIFY Ⅱ;002 試験(国際共同試験))11, 12, 13)
11)承認時評価資料(第Ⅲ相試験(MODIFY Ⅰ;001 試験))
12)承認時評価資料(第Ⅲ相試験(MODIFY Ⅱ;002 試験))
13)承認時評価資料(第Ⅲ相試験(MODIFY Ⅰ;001試験/MODIFY Ⅱ;002試験)併合解析)
目的:
001試験
標準治療抗菌薬併用下のジーンプラバ®+actoxumab*単回投与が、投与後12週間のCDI再発率を低下させるかを、ジーンプラバ®、actoxumab 又はプラセボ単回投与と比較し、さらに安全性 プロファイルを検討する。
002試験
標準治療抗菌薬併用下のジーンプラバ®+actoxumab又はジーンプラバ®単回投与が、投与後12週間のCDI再発率を低下させるか、プラセボ単回投与と比較し、さらに安全性プロファイルを検討する。
対象:
CDIの初回発症時又は再発時に経口抗菌薬の標準治療(メトロニダゾール、バンコマイシン又はフィダキソマイシン)を受けている18歳以上のCDI(下痢を呈し、かつC. difficileトキシン便検査が陽性)患者
方法:
001試験
被験者を1:1:1:1の比で4群〔ジーンプラバ®(10mg/kg)、actoxumab(10mg/kg)、ジーンプラバ®+actoxumab併用(各10mg/kg)及びプラセボ〕のいずれかに無作為に割り付けた(各群400例目標)。無作為割付けは、標準治療抗菌薬及び入院状態(入院又は外来)によって層別した。治験薬を単回静脈内投与し、有効性は投与後12週間、有害事象及び臨床検査は投与後4週間(重篤な有害事象は投与後12週間)まで検討した。
002試験
被験者(日本人を含む)を1:1:1の比で3群〔ジーンプラバ®(10mg/kg)、ジーンプラバ®+actoxumab併用(各10mg/kg)及びプラセボ〕のいずれかに無作為に割り付けた(各群400例目標)。無作為割付けは、標準治療抗菌薬及び入院状態(入院又は外来)によって層別した。治験薬を単回静脈内投与し、有効性は投与後12週間、有害事象及び臨床検査は投与後4週間(重篤な有害事象は投与後12週間)まで検討した。さらに、12ヵ月目までのCDI再発を検討するために、12週間の主試験を完了した被験者のうち約300例で9ヵ月間の延長試験を行った。
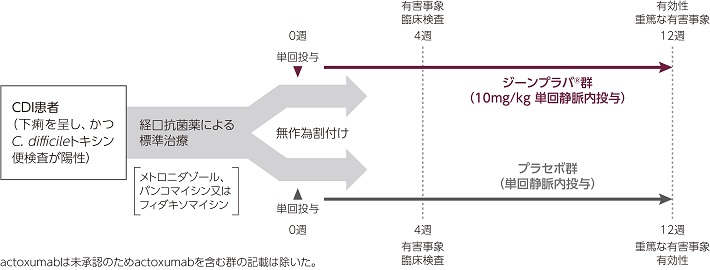
*未承認。本試験において、actoxumab単独群のCDI再発率はプラセボ群と同程度であったこと、またジーンプラバ®とactoxumab併用群においても、ジーンプラバ®単独群を上回るベネフィットが得られなかったことから、actoxumabの開発は中止された。本臨床試験成績については、actoxumabを含む群のデータを除外して紹介する。
評価項目:
【主要評価項目】最大の解析対象集団(FAS)における、治験薬投与後12週間までのCDI再発率
【副次評価項目】持続的治癒率、CDI再発リスクが高い部分集団(CDI既往歴あり、65歳以上、免疫不全状態の患者、重症のCDI)におけるCDI再発率
【探索的評価項目】CDI 再発までの期間
【安全性評価項目】臨床症状の有害事象、バイタルサイン、臨床検査値、心電図
解析方法:
001試験
中間解析において、ジーンプラバ®群及び/又はactoxumab群のCDI再発率がこれらの併用群と有意な差を認めた場合、該当する群の組入れを中止するというアダプティブデザインであった。
主要評価項目について、無作為割付け時点の標準治療抗菌薬及び入院状態を層別因子とするMiettinen and Nurminenの方法を用いて、階層手順により最初に併用群とプラセボ群を比較し(有意水準:片側1.25%)、有意差が認められた場合にジーンプラバ®群及びactoxumab群とプラセボ群との比較を行うこととしたが、中間解析においてactoxumab群の組入れが中止されたことから、ジーンプラバ®群とプラセボ群の比較を行った(有意水準:片側1.25%)。部分集団解析では層別因子で調整せずに、Miettinen and Nurminenの方法を用いて信頼区間を算出した。
002試験
主要評価項目について、無作為割付け時点の標準治療抗菌薬及び入院状態を層別因子とするMiettinen and Nurminenの方法を用いて、ジーンプラバ®群及び併用群とプラセボ群を比較した(有意水準:片側2.5%)。部分集団解析では層別因子で調整せずに、Miettinen and Nurminenの方法を用いて信頼区間を算出した。
日本人部分集団については、治験実施計画書に記載されていないが、CDI再発率及び持続的治癒率を算出し、ジーンプラバ®群とプラセボ群の比較、検討を行い、評価資料として承認時に評価された。
2試験の併合解析
併合解析は、大規模な被験者コホートを用いて治験薬投与後12週間のジーンプラバ®の有効性を検討し、個々の試験(001試験及び002試験)の結果を確認して、精度を向上させることを目的とした。有効性(主要評価項目及び副次評価項目)の併合解析では、正式な仮説検定ではなく、多重性を考慮せず、各評価項目での治療効果の尺度として、Miettinen and Nurminenの方法を用いて信頼区間及び名目上のp値(片側)を算出した。なお、併用した標準治療抗菌薬別のCDI再発率をサブグループ解析し算出した。
CDI再発:初回CDIが臨床的治癒に至った後、新たな下痢(24時間以内に3回以上の軟便)を発現し、それに伴うC. difficileトキシン便検査が陽性となった場合
臨床的治癒:初回CDIに対して標準治療を受けた期間が14日間以下であり、かつ、標準抗菌薬治療の完了直後2日間連続して下痢がない(24時間当たりの軟便回数が2回以下)場合
持続的治癒:初回CDIが臨床的治癒に至り、かつ、治験薬投与後12週目までCDI再発がない場合
有効性(001試験及び002試験)
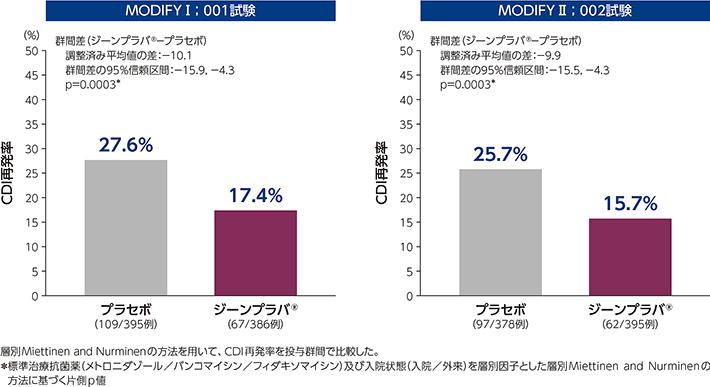
1)CDI再発率(主要評価項目、FAS)
投与後12週間のCDI再発率は、ジーンプラバ®群とプラセボ群の比較において、いずれの試験でも統計学的に有意な差が認められました(いずれもp=0.0003*)。
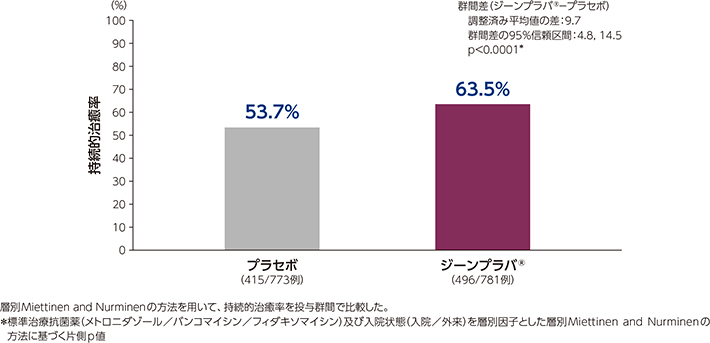
2)持続的治癒率(001試験/002試験併合解析:副次評価項目)
併合解析におけるジーンプラバ®群及びプラセボ群の持続的治癒率は、それぞれ63.5%及び53.7%であり、ジーンプラバ®群はプラセボ群に対して有意な差が認められました(p<0.0001*)。
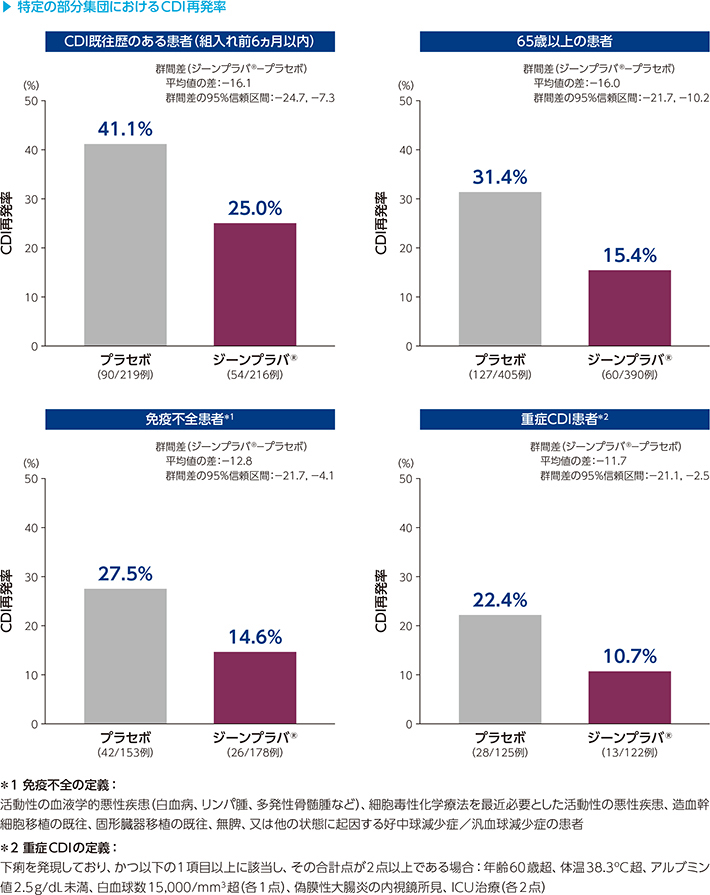
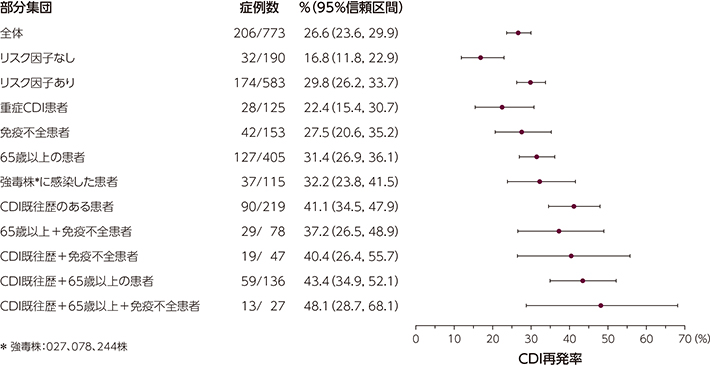
3)CDI再発リスクが高い部分集団におけるCDI再発率
(001試験/002試験併合解析:副次評価項目)
<参考>プラセボ群におけるCDI再発リスク因子別のCDI再発率
本試験結果は、事後解析であるが、CDI再発に対するリスク因子を明らかにするために実施しました。
プラセボ群において、リスク因子がない患者の再発率は16.8%、リスク因子がある患者では29.8%でした。
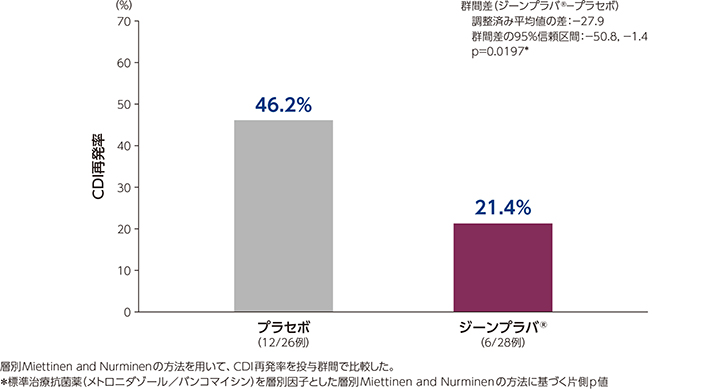
有効性(002試験における日本人に対するサブグループ解析)
本結果は評価資料として承認時に評価されたサブグループ解析です。
1)CDI再発率(主要評価項目、FAS)
日本人集団におけるジーンプラバ®群及びプラセボ群のCDI再発率は、それぞれ21.4%及び46.2%でした。
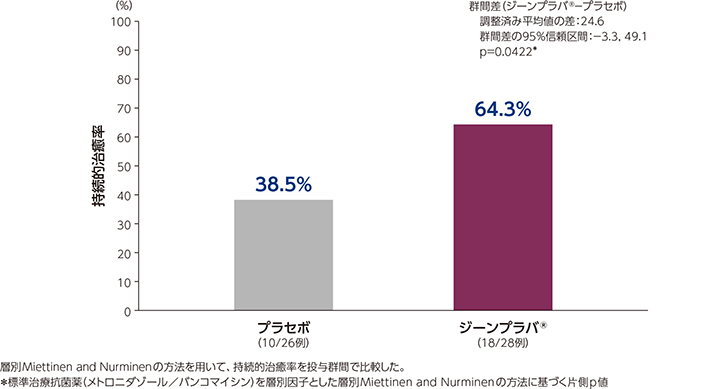
2)持続的治癒率(副次評価項目)
ジーンプラバ®群及びプラセボ群の持続的治癒率は、それぞれ64.3%及び38.5%でした。
安全性(001試験及び002試験)
001試験における副作用発現率
投与後4週間の副作用発現率はジーンプラバ®群8.2%(32/390例)、プラセボ群5.0%(20/400例)でした。発現率が1%以上であった副作用は、ジーンプラバ®群の浮動性めまい1.0%(4/390例)でした。
投与後12週間の重篤な副作用はジーンプラバ®群で4例(1.0%)、プラセボ群で1例(0.3%)でした。重篤な副作用の内容は、ジーンプラバ®群では下痢が1例、心室性頻脈性不整脈が1例、血尿が1例、敗血症及び脳出血が1例であり、プラセボ群では扁平上皮癌が1例でした。敗血症及び脳出血を発現した被験者は死亡しました。投与中止に至った副作用はジーンプラバ®群で1例に認められ(上記の重篤な副作用である心室性頻脈性不整脈)、副作用による死亡例はジーンプラバ®群で1例(敗血症及び脳出血)でした。
002試験における副作用発現率
投与後4週間の副作用発現率はジーンプラバ®群6.8%(27/396例)、プラセボ群6.8%(26/381例)でした。発現率が1%以上であった副作用は、ジーンプラバ®群の悪心1.3%(5/396例)及び疲労1.0%(4/396例)並びにプラセボ群の疲労1.0%(4/381例)でした。
投与後12週間の重篤な副作用はジーンプラバ®群では認められず、プラセボ群で1例(0.3%)に肺塞栓症がみられました。本試験において投与中止に至った副作用及び副作用による死亡例はありませんでした。
002試験の日本人集団における副作用発現率
投与後4週間の副作用発現率はジーンプラバ®群20.7%(6/29例)、プラセボ群23.1%(6/26例)でした。2例以上に発現した副作用は、ジーンプラバ®群のアスパラギン酸アミノトランスフェラーゼ増加6.9%(2/29例)であり、プラセボ群では認められませんでした。
投与後12週間の重篤な副作用、投与中止に至った副作用及び副作用による死亡例はありませんでした。