DRIVE-SHIFT試験
本剤は海外での臨床試験成績をもとに承認されたため、国内での臨床試験は実施していません。
DRIVE-SHIFT試験で投与したドラビリン/ラミブジン/テノホビルジソプロキシルフマル酸塩の配合剤(DOR/3TC/TDF)※は国内未承認薬ですが、承認時評価資料であることからデータを掲載しています。これらの各成分の国内承認用法及び用量については本ページ注釈又は各製品電子添文をご参照ください。
DRIVE-SHIFT試験
治療経験があるHIV-1感染症患者を対象とした海外第Ⅲ相非盲検比較試験(海外データ)1)2)
1)Johnson M, et al. J Acquir Immune Defic Syndr 2019; 81(4):463-472
(利益相反:Merck & Co., Inc., Rahway, N.J., U.S.A.が資金提供。共著者のZhou、Morais、Kumar、Sklar、Hanna、
Hwang、GreavesはMerck & Co., Inc., Rahway, N.J., U.S.A.の社員。)
2)承認時評価資料(海外第Ⅲ相試験:DRIVE-SHIFT試験)
[目的]
抗HIV薬投与(ベースライン治療)で安定したウイルス学的抑制が得られているHIV-1感染症患者を対象、にDOR/3TC/TDF※へ切り替えた場合のウイルス学的有効性をベースライン治療DSGと比較検討する。
[試験デザイン]
多施設共同、無作為化、非盲検、実薬対照第Ⅲ相、非劣性検証試験

[対象]
18歳以上で、抗HIV薬投与で6ヵ月以上ウイルス学的抑制(HIV-1 RNA量<40copies/mL)が維持されている成人HIV-1感染症患者670例(試験薬投与例)
[方法]
①リトナビル又はコビシスタットを併用したプロテアーゼ阻害剤(PI)、もしくは②コビシスタットを併用したエルビテグラビル、又は③非ヌクレオシド系逆転写酵素阻害剤(NNRTI)に、ヌクレオシド系逆転写酵素阻害剤(NRTI)2剤を併用した抗HIV薬投与(ベースライン治療)で安定している患者を、DOR/3TC/TDF※を1日1回経口投与にすぐに切り替えた群(ISG群)と、24週までベースライン治療をDSGし、24週時にDOR/3TC/TDFに切り替えた群(DSG群)に2:1の割合で無作為に割付け、48週間追跡した。
[評価項目]
主要評価項目[検証的解析項目]:ISG群48週時及びDSG群24週時におけるHIV-1 RNA量<50copies/mLの患者の割合
副次評価項目:ISG群24週時及びDSG群24週時におけるHIV-1 RNA量<50copies/mL/≥50copies/mLの患者の割合、ISG群48週時及びDSG群24週時におけるHIV-1 RNA量≥50copies/mLの患者の割合、ISG群48週時及びDSG群24週時とISG群24週時及びDSG群24週時におけるHIV-1 RNA量<40copies/mLの患者の割合、免疫学的有効性(CD4陽性リンパ球数のベースラインからの変化量)等
追加評価項目:抗ウイルス薬耐性の発現 等
安全性評価項目:副作用、ベースライン治療がリトナビルを併用したPIベースの患者におけるISG群24週時及びDSG群24週時のLDLコレステロール及びNon-HDLコレステロールのベースラインからの変化量 等
[解析計画]
有効率の群間差の95%信頼区間(95%CI)は、各投与群の層(ベースライン治療別)あたりの症例数の調和平均により重み付けした差を用いた層補正Mantel-Haenszel法で算出した。CD4陽性リンパ球数の平均変化量の差の95%CIは、t分布に基づいて求めた。主要評価項目について、群間差(ISG群48週時-DSG群24週時)の95%CIの下限値が-8%を上回る場合に、ISG群のDSG群に対する非劣性が検証されるとした。非劣性が検証され、かつ95%CIの下限値が0%を上回る場合に、ISG群のDSG群に対する優越性が検証されるとした。また、副次評価項目である24週時のISG群及びDSG群のHIV-1 RNA量<50copies/mLの患者の割合について、ベースライン時の患者背景因子別にサブグループ解析を実施した。
※DOR/3TC/TDFは、ドラビリン100mg/ラミブジン300mg/テノホビル ジソプロキシルフマル酸塩300mgを含有する製剤で、国内未承認です。
各成分の国内承認⽤法及び⽤量(各製品電子添文より):
・ドラビリン:通常、成人にはドラビリンとして100mgを1⽇1回経口投与する。本剤は、食事の有無にかかわらず投与できる。投与に際しては、必ず他の抗HIV薬と併⽤すること。
・ラミブジン:通常、成人には他の抗HIV薬と併⽤して、ラミブジンとして1⽇量300mgを1⽇1回又は2回(150mg×2)に分けて経口投与する。なお、年齢、体重、症状により適宜増減する。
・テノホビル ジソプロキシルフマル酸塩:通常、成人にはテノホビル ジソプロキシルフマル酸塩として1回300mg(テノホビル ジソプロキシルとして245mg)を1⽇1回経口投与する。なお、投与に際しては必ず他の抗HIV薬と併⽤すること。
有効性
■ ウイルス学的有効性
(主要評価時点:ISG群48週時 vs DSG群24週時、副次評価時点:ISG群24週時 vs DSG群24週時)
主要評価項目であるISG群48週時及びDSG群24週時におけるHIV-1 RNA量<50copies/mLの患者の割合(検証的解析項目)が、ISG群48週時90.8%、DSG群24週時94.6%であり、群間差-3.8%(95%CI:-7.9, 0.3、Mantel-Haenszel法)の95%CIの下限値が-8%を上回ったため、ISG群48週時のDSG群24週時に対する非劣性が検証されました。一方、95%CIの下限値が0%を上回らなかったため、優越性は示されませんでした。
ウイルス学的有効性a,1)2)
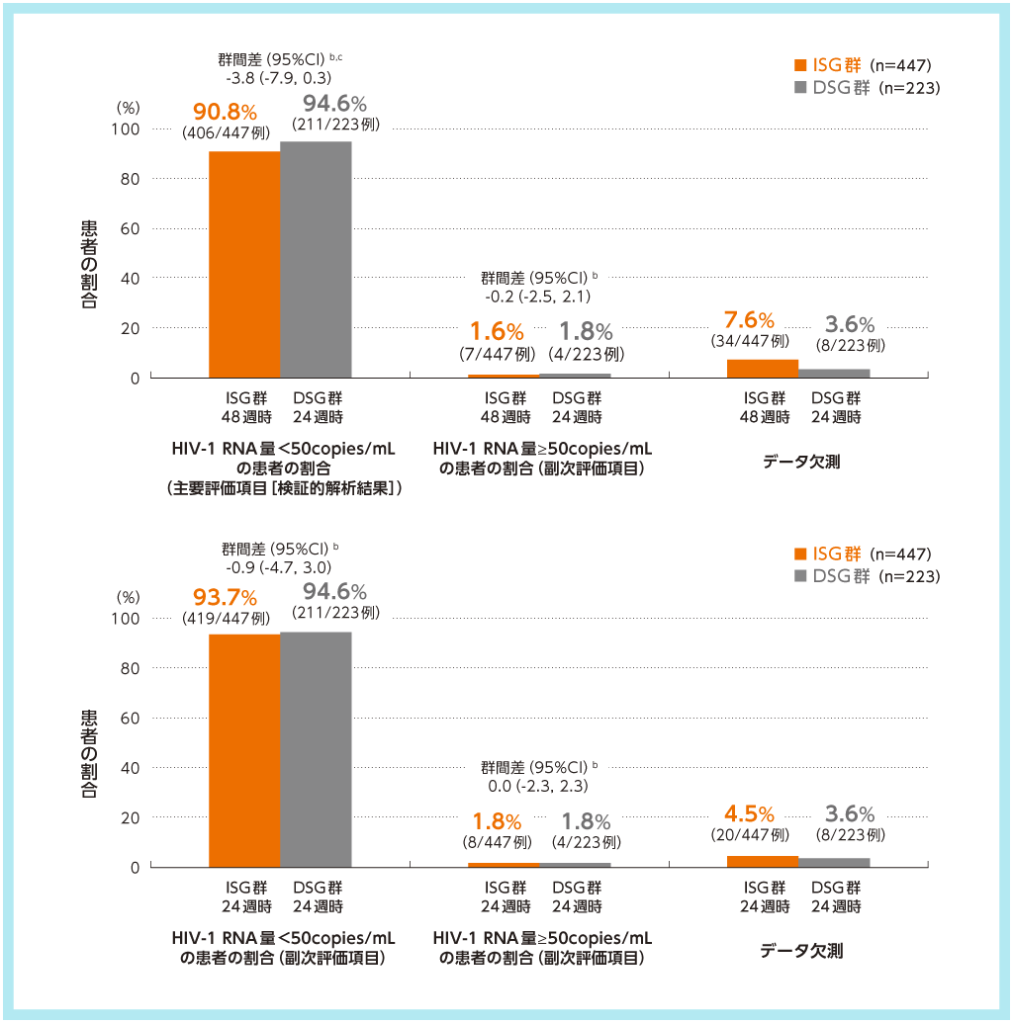
ISG群:試験開始時にDOR/3TC/TDFに切り替えて48週間投与
DSG群:ベースライン治療を24週間DSG後にDOR/3TC/TDFに切り替えて24週間投与
a:FDA Snapshot法:理由を問わず、試験の中止を含めて全てのデータ欠損を治療失敗として扱った。
b:投与群間差(ISG群-DSG群)の95%CIは、各投与群の層(ベースライン治療:リトナビルもしくはコビシスタットを併用したPIベース、コビシスタットを併用したEVGベース又はNNRTIベース)あたりの症例数の調和平均により重み付けした差を用いた層補正Mantel-Haenszel法により算出した。
c:主要評価項目について、HIV-1 RNA量<50copies/mLを達成した患者の割合の投与群間差の95%CIの下限値が-8%を上回る場合に、ISG群のDSG群に対する非劣性が検証されるとした。非劣性が検証され、かつ95%CIの下限値が0%を上回る場合に、ISG群のDSG群に対する優越性が検証されるとした。
ウイルス学的有効性及び免疫学的有効性2)
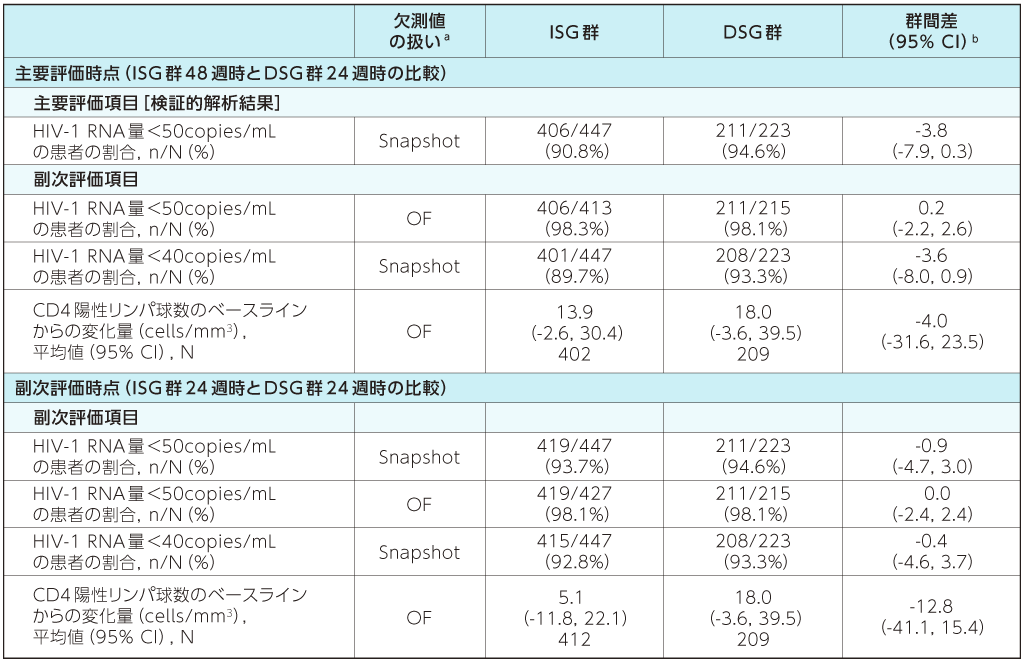
ISG群:試験開始時にDOR/3TC/TDFに切り替えて48週間投与
DSG群:ベースライン治療を24週間DSG後にDOR/3TC/TDFにすぐ切り替えて24週間投与
a:Snapshot:FDA Snapshot法:理由を問わず、試験の中止を含めて全てのデータ欠損を治療失敗として扱った。
OF:Observed Failure法:効果不十分のために試験薬投与を早期に中止し、欠測値(投与期間中に断続的に生じる欠測値を除く)のある症例を治療失敗例として扱った。
b:群間差の95%CIは、各投与群の層(ベースライン治療:リトナビルもしくはコビシスタットを併用したPIベース、コビシスタットを併用したEVGベース又はNNRTIベース)あたりの症例数の調和平均により重み付けした差を用いた層補正Mantel-Haenszel法により算出した。CD4陽性リンパ球数の平均変化量の差の95%CIは、t分布に基づいて求めた。
安全性
■ 副作用
48週までのDOR/3TC/TDF投与群(ISG群0~48週+DSG群24~48週、n=656)の副作用発現率は19.7%(129/656例)であり、主な副作用はALT上昇14例(2.1%)、頭痛12例(1.8%)でした。
DOR/3TC/TDF投与群で48週までに発現した重篤な副作用は、5例でリパーゼ増加2件、ALT増加、AST増加、アミラーゼ増加、うつ病及び腎不全の各1件でした。DOR/3TC/TDF投与群で48週までに試験薬の投与中止に至った副作用は、13例でリパーゼ増加、ALT増加及びAST増加の各2件、全身性浮腫、筋肉痛、斑状皮疹、腎臓痛、記憶障害、疲労、睡眠障害、うつ病、食欲減退、アミラーゼ増加、腎不全、記憶障害及び肝炎の各1件でした。
24週までのDSG群の副作用発現率は2.2%(5/223例)で、主なものは頭痛1例(0.4%)でした。
24週までのDSG群に重篤な副作用ならびに試験薬の投与中止に至った副作用の発現は認められませんでした。
48週間の試験期間中に、いずれの投与群においても死亡は認められませんでした。
■〔参考情報〕脂質プロファイルへの影響
スクリーニング時のベースライン治療がリトナビルを併用したPIベースの患者では、24週時の空腹時LDLコレステロール及びNon-HDLコレステロールのベースラインからの変化量は、ISG群で-16.5及び-24.7mg/dL、DSG群でそれぞれ-1.9及び-1.3mg/dLでした。
24週時における空腹時脂質のベースラインからの変化量(安全性評価項目ベースライン治療がリトナビルを併用したPIベースの患者)2)
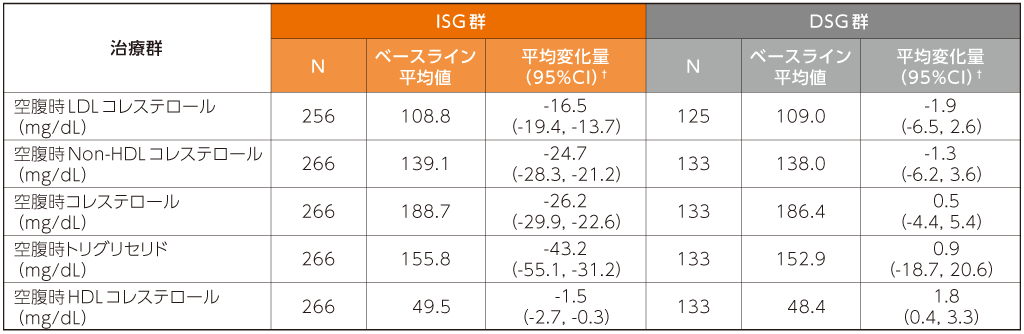
ISG群:試験開始時にDOR/3TC/TDFに切り替えて48週間投与
DSG群:ベースライン治療を24週間DSG後にDOR/3TC/TDFに切り替えて24週間投与
欠測値は、Last Observation Carried Forward(LOCF)法を用いて補完した。
†:群内の95%CIはt分布に基づいた。
N:スクリーニング時の治療レジメンがリトナビルを併用したPIベースで、ベースライン後の検査結果が1つ以上得られた患者の数