KEYNOTE-522試験(高リスク早期)
<高リスク早期トリプルネガティブ乳癌>
トリプルネガティブ乳癌:国際共同第Ⅲ相試験<KEYNOTE-522試験>
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-522試験)
Schmid P et al. N Engl J Med 2020; 382: 810-821
Schmid P et al. N Engl J Med 2022; 386: 556-567
Schmid P et al. N Engl J Med 2024; 391: 1981-1991
Schmid P et al. N Engl J Med 2024; 391: 1981-1991. Supplementary Appendix
社内資料:国際共同第Ⅲ相試験(KEYNOTE-522試験)(安全性データ)
本試験はMSD社の資金提供により行われた。Peter SchmidはMSD社からコンサルタント料を受領している。また、著者のうち、Yu Ding、Konstantinos Tryfonidis、Vassiliki Karantza、Gursel Aktan、Liyi Jia、Jing Zhaoは同社の社員である。その他の著者にMSD社よりコンサルタント料、講演料などを受領している者が含まれる。
試験概要
【 目的 】
高リスクの早期トリプルネガティブ乳癌患者における、キイトルーダ®+化学療法併用(術前薬物療法)及びキイトルーダ®単独(術後薬物療法)の有効性及び安全性を比較検討する。
【デザイン】
国際共同無作為化二重盲検第Ⅲ相試験[優越性試験]
[第1回中間解析結果(データカットオフ日:2018年9月24日)]
[第4回中間解析結果(データカットオフ日:2021年3月23日)]
[OS最終解析結果(データカットオフ日:2024年3月22日)]
【 対象 】
高リスクの早期トリプルネガティブ乳癌患者*11,174例(うち日本人患者76例を含む)
【 方法 】
キイトルーダ®群とプラセボ群に2:1の割合で無作為に割り付けた。
キイトルーダ®群:術前薬物療法①として、キイトルーダ®200㎎を3週間間隔(Q3W、21日を1サイクル)、パクリタキセル80mg/m2を1週間間隔(QW、21日を1サイクル)、カルボプラチン*2AUC5に相当する量をQ3W又はAUC1.5に相当する量をQWで4サイクル点滴静注後、術前薬物療法②として、キイトルーダ®200㎎をQ3W、ドキソルビシン塩酸塩*360mg/m2をQ3W又はエピルビシン塩酸塩*390mg/m2をQ3W、シクロホスファミド600mg/m2(無水物換算)をQ3Wで4サイクル点滴静注した後に根治手術*4を行った。続けて術後薬物療法としてキイトルーダ®200mgをQ3Wで9サイクル点滴静注した。
プラセボ群:術前薬物療法①として、プラセボをQ3W、パクリタキセル80mg/m2をQW、カルボプラチンAUC5に相当する量をQ3W又はAUC1.5に相当する量をQWで4サイクル点滴静注後、術前薬物療法②として、プラセボをQ3W、ドキソルビシン塩酸塩60mg/m2をQ3W又はエピルビシン塩酸塩90mg/m2をQ3W、シクロホスファミド600mg/m2(無水物換算)をQ3Wで4サイクル点滴静注した後に根治手術を行った。続けて術後薬物療法としてプラセボをQ3Wで9サイクル点滴静注した。
投与完了(キイトルーダ®又はプラセボは17サイクル)、疾患進行又は再発、許容できない有害事象の発現等が生じるまで継続した。プラセボからキイトルーダ®へのクロスオーバーは許容しなかった。
a:パクリタキセル80mg/m2 QW 点滴静注(各サイクルの1、8、15日目に投与、21日を1サイクル)
b:カルボプラチンAUC5に相当する量 Q3W 点滴静注(各サイクルの1日目に投与、21日を1サイクル)又はAUC1.5に相当する量 QW 点滴静注(各サイクルの1、8、15日目に投与、21日を1サイクル)
c:ドキソルビシン塩酸塩60mg/m2 Q3W 点滴静注(各サイクルの1日目に投与、21日を1サイクル)
d:シクロホスファミド600mg/m2(無水物換算) Q3W 点滴静注(各サイクルの1日目に投与、21日を1サイクル)
e:エピルビシン塩酸塩90mg/m2 Q3W 点滴静注(各サイクルの1日目に投与、21日を1サイクル)
【評価項目】
主要評価項目:ITT(intention-to-treat)集団*8における病理学的完全奏効(pathological complete response:pCR)率(ypT0/Tis ypN0※1)※2、無イベント生存期間(event-free survival:EFS)*9、※2
副次評価項目:ITT集団におけるpCR率(ypT0 ypN0※1)、pCR率(ypT0/Tis※1)、全生存期間(overall survival:OS)※2、安全性解析対象集団*10における安全性及び忍容性 等
探索的評価項目:FAS(full analysis set)集団*11における無遠隔再発生存期間(distant recurrence-free survival:DRFS)*12、ITT集団におけるpCR(ypT0/Tis ypN0)達成別のEFS、根治手術時の乳房温存手術の割合、残存腫瘍量*13 等
※1 各pCRの定義
ypT0/Tis ypN0:乳房内の浸潤巣とリンパ節転移がみられないもので、非浸潤巣の有無は問わないもの
ypT0 ypN0:乳房内の浸潤巣、非浸潤巣、リンパ節転移いずれもみられないもの
ypT0/Tis:乳房内の浸潤巣がみられないもので、非浸潤巣とリンパ節転移の有無は問わないもの
※2 検証的解析項目
【判定基準】
pCRは根治手術の際に、治験実施医療機関の病理医がAJCC Breast Cancer Stagingに基づき判定した。EFSのイベントは治験担当医師が判定した。
【解析計画】
解析対象集団:対照群との有効性の比較はITT集団、安全性は安全性解析対象集団を解析対象とした。DRFSの解析対象はFAS集団とした。
有効性評価の統計手法:pCR率の群間差及びその95%CIは、例数で重み付けした層別Miettinen and Nurminen法により評価した。EFS及びOSは、Kaplan-Meier法を用いて生存曲線を推定し、群間比較には層別ログランク検定を用いた。EFSのハザード比及び95%CIは、投与群を共変量とした層別Cox比例ハザードモデルを用いて算出した。OSについては、比例ハザード性が成立しなかったため区間ごとのハザード比を算出し、全体の効果として一つの数値に要約するため、イベント数で重み付けした平均ハザード比を算出した。リンパ節転移の有無、腫瘍サイズ、カルボプラチンの投与スケジュール、閉経状態、年齢、地域、ECOG PS、HER2発現、乳酸脱水素酵素(LDH)、ステージ*14について、pCR率(ypT0/Tis ypN0)及びEFSは、Miettinen and Nurminen法及びCox比例ハザードモデルを用いて、OSは、5年生存率の差を用いて部分集団解析を実施した。PD-L1発現別の解析について、pCR率は層別Miettinen and Nurminen法、EFSのハザード比及び95%CIは投与群を共変量とした層別Cox比例ハザードモデル、OSは5年生存率の差を用いて算出した。DRFSはEFSと同様の統計手法で評価した。pCR(ypT0/Tis ypN0)達成別のEFSについてはKaplan-Meier法を用いて生存曲線を推定し、ハザード比及び95%CIは投与群を共変量としたCox比例ハザードモデルを用いて算出した。日本人集団については、ITT集団と同様の統計手法により解析したが、層での調整は行わなかった。
多重性の調整:本試験では、7回の中間解析を事前に計画し、試験全体の有意水準を片側2.5%に厳密に制御した。pCR(ypT0/Tis ypN0)は2回(1回目及び2回目)、EFSは6回(2回目以降)の中間解析及び最終解析を実施することとした。また、EFSの優越性が示された場合はOSを検定することとした。pCR(ypT0/Tis ypN0)、EFS及びOSの多重性の調整は、Maurer and Bretzのgraphical approachを用いた。中間解析と最終解析における有意水準の配分には、α消費関数を用いた。

※3:仮説2の優越性が検証され、有意水準が再配分された場合に検定
*1 中央検査機関でトリプルネガティブ乳癌と確認された患者
*2 治験担当医師の判断でいずれかの投与スケジュールを選択
*3 治験担当医師の判断でいずれかの薬剤を選択
*4 術前薬物療法期の完了又は早期中止の約3~6週間後に手術を実施
*5 放射線評価及び/又は臨床評価に基づき治験担当医師が評価
*6 Eastern Cooperative Oncology Group performance status
*7 術後薬物療法は根治手術後約30~60日以内に開始する
*8 無作為化されたすべての患者
*9 無作為化から疾患進行による根治手術不能、局所再発又は遠隔再発、二次がん、あらゆる原因による死亡のいずれかの事象が最初に記録された日までの期間
*10 無作為化され、治療(治験薬投与又は手術)を1回以上受けたすべての患者
*11 無作為化され、根治手術を実施したすべての患者(すなわち、術前薬物療法中に遠隔増悪した患者、手術未施行の患者又は直近の手術時に切除断端陽性であった患者は除外された)
*12 根治手術から治験担当医師が評価する遠隔転移発生までの期間
*13 根治手術時に治験実施医療機関の病理医の評価による乳房又はリンパ節の残存腫瘍
*14 ステージ別の部分集団解析はEFSのみ実施した
パクリタキセルの用法及び用量は以下のとおりです。
【用法及び用量】(抜粋)
乳癌にはA法又はB法を使用する。
A法:通常、成人にはパクリタキセルとして、1日1回210mg/m2(体表面積)を3時間かけて点滴静注し、少なくとも3週間休薬する。これを1クールとして、投与を繰り返す。
B法:通常、成人にはパクリタキセルとして、1日1回100mg/m2(体表面積)を1時間かけて点滴静注し、週1回投与を6週連続し、少なくとも2週間休薬する。これを1クールとして、投与を繰り返す。
なお、投与量は、患者の状態により適宜減量する。
エピルビシン塩酸塩の用法及び用量は以下のとおりです。
【用法及び用量】(抜粋)
乳癌(手術可能例における術前、あるいは術後化学療法)に対する他の抗悪性腫瘍剤との併用療法の場合
・シクロホスファミド水和物との併用において、標準的なエピルビシン塩酸塩の投与量及び投与方法は、エピルビシン塩酸塩として100mg(力価)/m2(体表面積)を1日1回静脈内に投与後、20日間休薬する。これを1クールとし、通常4~6クール反復する。
・シクロホスファミド水和物、フルオロウラシルとの併用において、標準的なエピルビシン塩酸塩の投与量及び投与方法は、エピルビシン塩酸塩として100mg(力価)/m2(体表面積)を1日1回静脈内に投与後、20日間休薬する。これを1クールとし、通常4~6クール反復する。
なお、投与量は年齢、症状により適宜減量する。
患者背景(ITT集団)


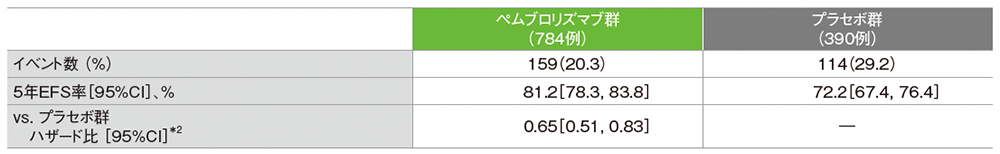
主要評価項目:優越性試験(検証的解析結果)
病理学的完全奏効率:pCR率(ypT0/Tis ypN0)*1
- pCR率は、キイトルーダ®群で64.8%(95%CI:59.9, 69.5)、プラセボ群で51.2%(95%CI:44.1, 58.3)であり、キイトルーダ®群はプラセボ群に対してpCR率を有意に改善しました(推定群間差:13.6%、95%CI:5.4, 21.8、p=0.00055、層別Miettinen and Nurminen法[片側]、有意水準α=0.003、検証的解析結果)。
■病理学的完全奏効率(pCR率: ypT0/Tis ypN0)(ITT集団)

*1 第1回中間解析時点でpCR率の解析対象であった、無作為化された最初の602例で解析を実施
*2 無作為化に用いた層別因子[リンパ節転移(あり、なし)、腫瘍サイズ(T1又はT2、T3又はT4)、カルボプラチンの投与スケジュール(Q3W、QW)]を層別因子とした層別Miettinen and Nurminen法[片側]に基づく(検証的解析結果)
(第1回中間解析、データカットオフ日:2018年9月24日)

主要評価項目:優越性試験(検証的解析結果)
無イベント生存期間:EFS*1
- プラセボ群に対するキイトルーダ®群のハザード比は0.63(95%CI:0.48,0.82)であり、EFSを有意に延長しました(p=0.0003093、層別ログランク検定[片側]、有意水準α=0.00516941、検証的解析結果)。
■無イベント生存期間(EFS)のKaplan-Meier曲線(ITT集団)

*1 無作為化から疾患進行による根治手術不能、局所再発又は遠隔再発、二次がん、あらゆる原因による死亡のいずれかの事象が最初に記録された日までの期間と定義。治験担当医師が評価
*2 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*3 投与群を共変量とし、無作為化に用いた層別因子[リンパ節転移(あり、なし)、腫瘍サイズ(T1又はT2、T3又はT4)、カルボプラチンの投与スケジュール(Q3W、QW)]を層別因子とした層別Cox比例ハザードモデルに基づく
*4 無作為化に用いた層別因子を層別因子とした層別ログランク検定[片側](検証的解析結果)
(第4回中間解析、データカットオフ日:2021年3月23日)
■無イベント生存期間(EFS)のイベントの要約(ITT集団)

(第4回中間解析、データカットオフ日:2021年3月23日)
サブグループ解析
部分集団因子別の無イベント生存期間:EFS
■無イベント生存期間(EFS)のハザード比のフォレストプロット(ITT集団)

* 全例及びPD-L1発現別のサブグループは投与群を共変量とし、無作為化に用いた層別因子[リンパ節転移(あり、なし)、腫瘍サイズ(T1又はT2、T3又はT4)、カルボプラチンの投与スケジュール(QW、Q3W)]を層別因子とした層別Cox比例ハザードモデル、その他のサブグループはCox比例ハザードモデルに基づく
(第4回中間解析、データカットオフ日:2021年3月23日)
探索的評価項目 サブグループ解析
病理学的完全奏効(pCR: ypT0/Tis ypN0)達成別の無イベント生存期間:EFS
術前薬物療法でpCRを達成したキイトルーダ®群の494例とプラセボ群の217例、non-pCRであったキイトルーダ®群の290例とプラセボ群の173例におけるEFSを探索的に検討しました。本解析は無作為化割付けによる比較ではないため、結果の解釈には注意が必要です。
- プラセボ群に対するキイトルーダ®群のハザード比は、pCR例で0.73(95%CI:0.39, 1.36)、non-pCR例0.70(95%CI:0.52, 0.95)でした。
- pCR例における36ヵ月時点のEFS率は、キイトルーダ®群で94.4%(95%CI:91.9, 96.2)、プラセボ群で92.5%(95%CI:88.1, 95.3)でした。non-pCR例における36ヵ月時点のEFS率は、キイトルーダ®群で67.4%(95%CI:61.6, 72.5)、プラセボ群で56.8%(95%CI:49.0, 63.9)でした。
■病理学的完全奏効(pCR: ypT0/Tis ypN0)達成別の無イベント生存期間(EFS)のKaplan-Meier曲線(ITT集団)

*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量としたCox比例ハザードモデルに基づく
(第4回中間解析、データカットオフ日:2021年3月23日)

主要評価項目:優越性試験
無イベント生存期間:EFS*1
掲載誌の規定に従い、最終解析の有効性は薬剤名(キイトルーダ®)を一般名(ペムブロリズマブ)で記載しています。
■ 無イベント生存期間(EFS)のKaplan-Meier曲線(ITT集団)
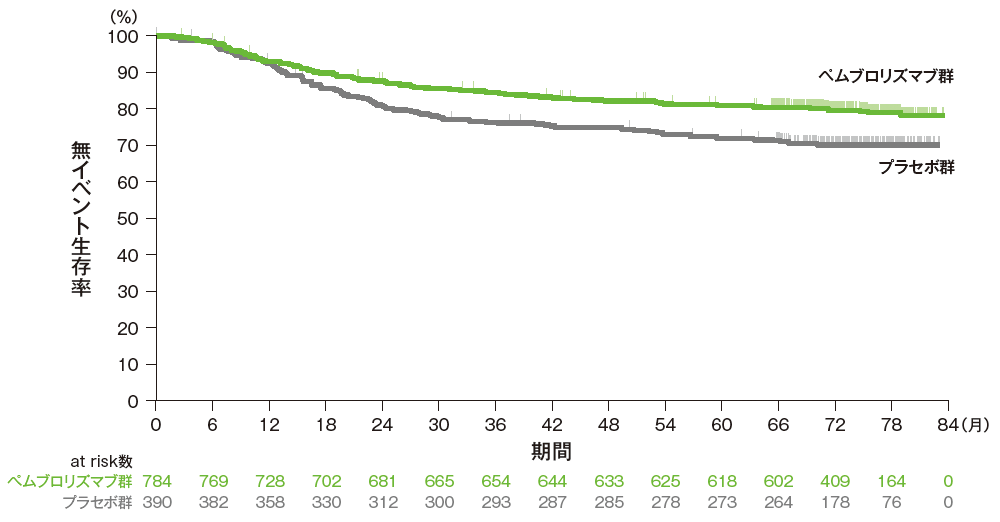
Adapted with permission from Overall Survival with Pembrolizumab in Early-Stage Triple-Negative Breast Cancer,
Schmid P et al, N Engl J Med, 2024; 391: 1981-1991.
Copyright ©2024 Massachusetts Medical Society. Translated with permission.
*1: 無作為化から疾患進行による根治手術不能、局所再発又は遠隔再発、二次がん、あらゆる原因による死亡のいずれかの事象が最初に記録された日までの期間と定義。治験担当医師が評価
*2: 投与群を共変量とし、無作為化に用いた層別因子[リンパ節転移(あり、なし)、腫瘍サイズ(T1又はT2、T3又はT4)、カルボプラチンの投与スケジュール(Q3W、QW)]を層別因子とした層別Cox比例ハザードモデルに基づく
(OS最終解析、データカットオフ日: 2024年3月22日、追跡期間中央値: 75.1ヵ月)
サブグループ解析
部分集団因子別の無イベント生存期間:EFS
掲載誌の規定に従い、最終解析の有効性は薬剤名(キイトルーダ® )を一般名(ペムブロリズマブ)で記載しています。
■ 無イベント生存期間(EFS)のハザード比のフォレストプロット(ITT集団)
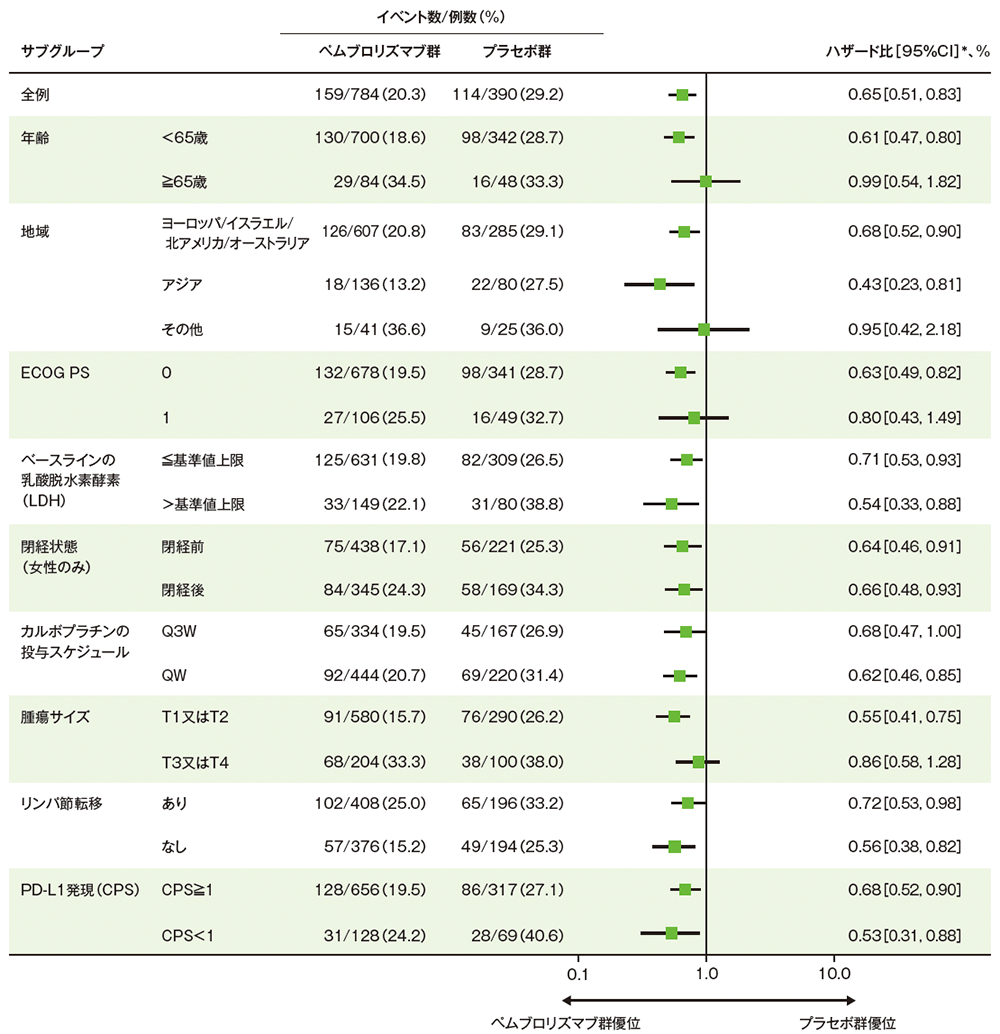
Adapted with permission from Overall Survival with Pembrolizumab in Early-Stage Triple-Negative Breast Cancer,
Schmid P et al, N Engl J Med, 2024; 391: 1981-1991.
Copyright ©2024 Massachusetts Medical Society. Translated with permission.
* 全例及びPD-L1発現別のサブグループは投与群を共変量とし、無作為化に用いた層別因子[リンパ節転移(あり、なし)、腫瘍サイズ(T1又はT2、T3又はT4)、カルボプラチンの投与スケジュール(QW、Q3W)]を層別因子とした層別Cox比例ハザードモデル、その他のサブグループはCox比例ハザードモデルに基づく
(OS最終解析、データカットオフ日: 2024年3月22日、追跡期間中央値: 75.1ヵ月)
副次評価項目:優越性試験(検証的解析結果)
全生存期間:OS
- プラセボ群に対するキイトルーダ®群のハザード比は0.72(95%CI: 0.51, 1.02)であり、第4回中間解析において優越性は検証されませんでした(p=0.0321377、層別ログランク検定[片側]、有意水準α=0.00085861、検証的解析結果)。
- 36ヵ月時点のOS率はキイトルーダ®群で89.7%(95%CI:87.3, 91.7)、プラセボ群で86.9%(95%CI:83.0, 89.9)でした。
■全生存期間(OS)のKaplan-Meier曲線(ITT集団)

*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量とし、無作為化に用いた層別因子[リンパ節転移(あり、なし)、腫瘍サイズ(T1又はT2、T3又はT4)、カルボプラチンの投与スケジュール(Q3W、QW)]を層別因子とした層別Cox比例ハザードモデルに基づく
*3 無作為化に用いた層別因子を層別因子とした層別ログランク検定[片側](検証的解析結果)
(第4回中間解析、データカットオフ日:2021年3月23日)
副次評価項目:優越性試験(検証的解析結果)
全生存期間:OS
掲載誌の規定に従い、最終解析の有効性は薬剤名(キイトルーダ® )を一般名(ペムブロリズマブ)で記載しています。
■ 全生存期間(OS)のKaplan-Meier曲線(ITT集団)
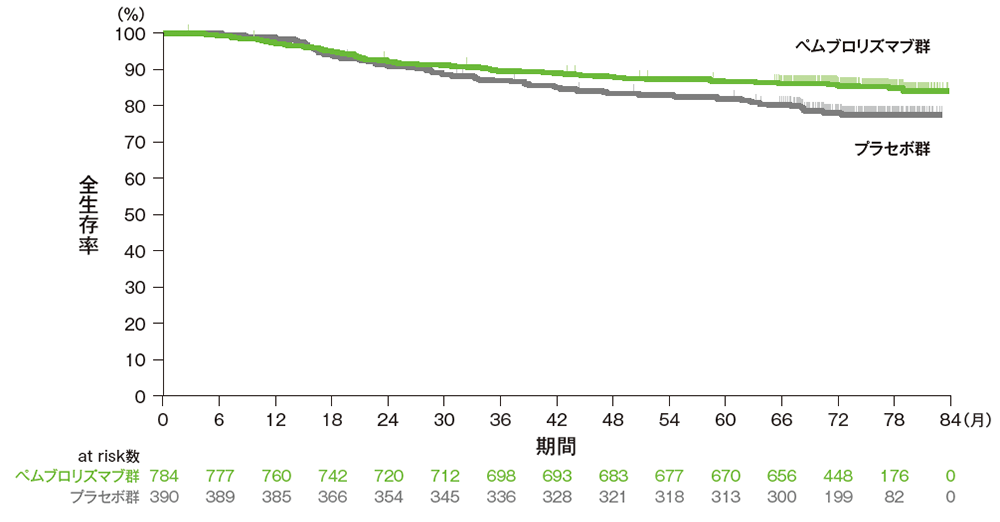
Adapted with permission from Overall Survival with Pembrolizumab in Early-Stage Triple-Negative Breast Cancer,
Schmid P et al, N Engl J Med, 2024; 391: 1981-1991.
Copyright ©2024 Massachusetts Medical Society. Translated with permission.

*1: 区間ごとのハザード比を算出し、全体の効果として一つの数値に要約するため、イベント数で重み付けした平均ハザード比を算出した
*2: 無作為化に用いた層別因子[リンパ節転移(あり、なし)、腫瘍サイズ(T1又はT2、T3又はT4)、カルボプラチンの投与スケジュール(Q3W、QW)]を層別因子とした層別ログランク検定[片側](検証的解析結果)
(OS最終解析、データカットオフ日:2024年3月22日、追跡期間中央値: 75.1ヵ月)
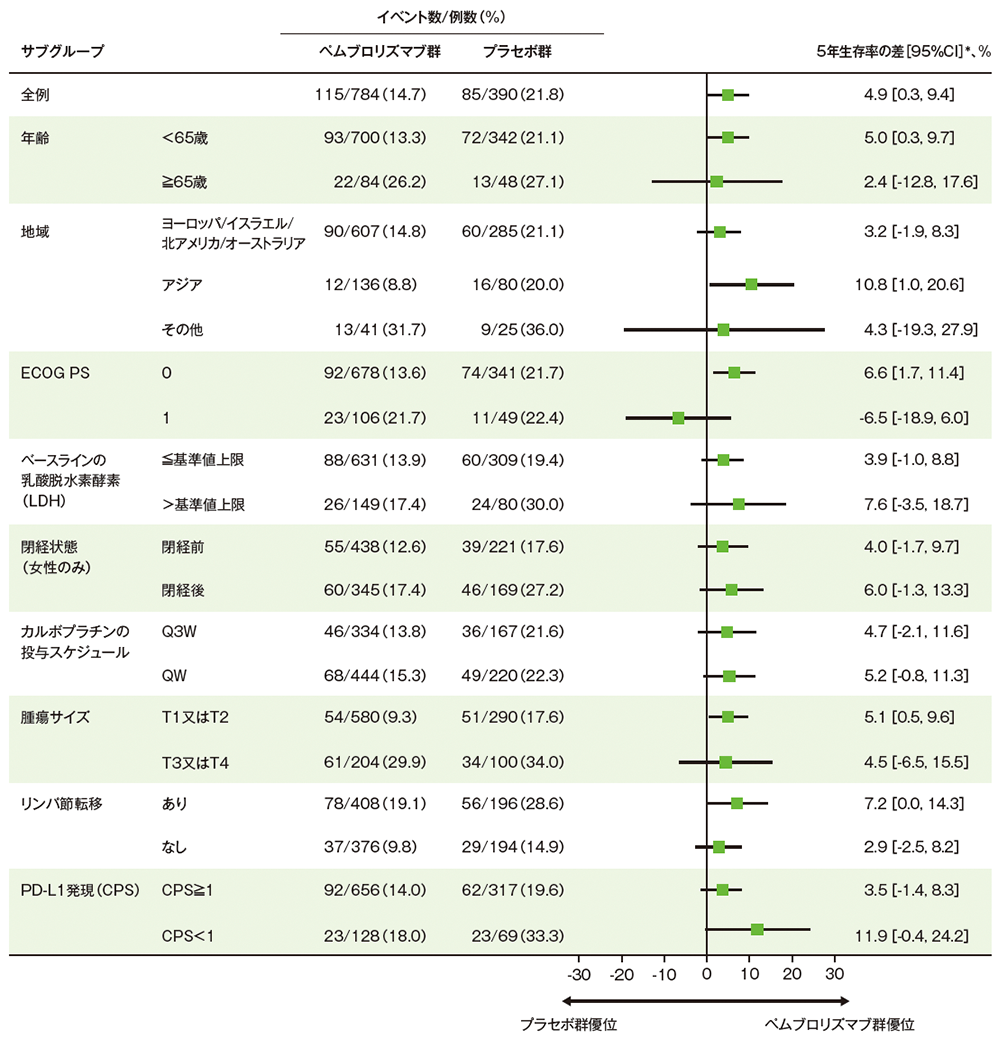
サブグループ解析
部分集団因子別の全生存期間:OS
掲載誌の規定に従い、最終解析の有効性は薬剤名(キイトルーダ® )を一般名(ペムブロリズマブ)で記載しています。
■ 全生存期間(OS)の5年生存率の差のフォレストプロット(ITT集団)
Adapted with permission from Overall Survival with Pembrolizumab in Early-Stage Triple-Negative Breast Cancer,
Schmid P et al, N Engl J Med, 2024; 391: 1981-1991.
Copyright ©2024 Massachusetts Medical Society. Translated with permission.
*両群における5年生存率のKaplan-Meier推定値がそれぞれ漸近的に正規分布に従うと仮定した場合の5年生存率の差及びその差の95%CIを算出(OS最終解析、データカットオフ日: 2024年3月22日、追跡期間中央値: 75.1ヵ月)
安全性(術前薬物療法期)
安全性解析対象集団の副作用及び免疫関連など特に注目すべき有害事象
■副作用
- キイトルーダ®群:773/783例(98.7%)
- プラセボ群:388/389例(99.7%)
■重篤な副作用
-
キイトルーダ®群:254例(32.4%)
内訳(1%以上):発熱性好中球減少症115例(14.7%)、発熱20例(2.6%)貧血19例(2.4%)、好中球減少症12例(1.5%)、汎血球減少症11例(1.4%)、副腎機能不全及び下垂体炎各8例(1.0%) -
プラセボ群:77例(19.8%)
内訳(1%以上):発熱性好中球減少症47例(12.1%)、貧血8例(2.1%)、汎血球減少症4例(1.0%)
■副作用による中止
-
キイトルーダ®群(いずれかの薬剤の中止):194例(24.8%)
キイトルーダ®の中止:115例(14.7%)
内訳(0.5%以上):アラニンアミノトランスフェラーゼ増加18例(2.3%)、アスパラギン酸アミノトランスフェラーゼ増加11例(1.4%)、発熱性好中球減少症及び下垂体炎各6例(0.8%)、肺臓炎5例(0.6%)、急性腎障害、副腎機能不全、大腸炎、肝毒性及び尿細管間質性腎炎各4例(0.5%)
化学療法の中止:130例(16.6%)
内訳(1%以上):好中球減少症15例(1.9%)、注入に伴う反応及び発熱性好中球減少症各10例(1.3%)、末梢性ニューロパチー8例(1.0%) -
プラセボ群(いずれかの薬剤の中止):49例(12.6%)
プラセボの中止:20例(5.1%)
内訳(0.5%以上):アラニンアミノトランスフェラーゼ増加4例(1.0%)、発熱性好中球減少症及び肺臓炎各2例(0.5%)
化学療法の中止:40例(10.3%)
内訳(1%以上):好中球減少症6例(1.5%)、末梢性ニューロパチー5例(1.3%)
■副作用による死亡
- キイトルーダ®群:2例 肺臓炎、多臓器機能不全症候群/敗血症各1例
- プラセボ群:1例 敗血症性ショック
■主な副作用(いずれかの群で10%以上に発現)(術前薬物療法期)

MedDRA/J v23.1、GradeはCTCAE v4.0
(第4回中間解析、データカットオフ日:2021年3月23日)
■主な免疫関連など特に注目すべき有害事象(いずれかの群で0.5%以上に発現)(術前薬物療法期)

MedDRA/J v23.1、GradeはCTCAE v4.0
ー:集計なし
同一症例がカテゴリー内の事象を複数発現した場合、カテゴリー別集計(太字)は1例とカウントしている
(第4回中間解析、データカットオフ日:2021年3月23日)
安全性(術後薬物療法期)
安全性解析対象集団の副作用及び免疫関連など特に注目すべき有害事象
■副作用
- キイトルーダ®群:316/588例(53.7%)
- プラセボ群:161/331例(48.6%)
■重篤な副作用
-
キイトルーダ®群:19例(3.2%)
内訳(2例以上):肺臓炎4例(0.7%)、肺塞栓症2例(0.3%) -
プラセボ群:2例(0.6%)
内訳(2例以上):認められなかった
■副作用による中止
-
キイトルーダ®群(キイトルーダ®の中止):25例(4.3%)
内訳(2例以上):関節痛4例(0.7%)、急性腎障害、心筋炎及び肺塞栓症各2例(0.3%) -
プラセボ群(プラセボの中止):6例(1.8%)
内訳(2例以上):認められなかった
■副作用による死亡
- キイトルーダ®群:2例 肺塞栓症、自己免疫性脳炎各1例
- プラセボ群:認められなかった
■主な副作用(いずれかの群で5%以上に発現)(術後薬物療法期)

MedDRA/J v23.1、GradeはCTCAE v4.0
(第4回中間解析、データカットオフ日:2021年3月23日)
■主な免疫関連など特に注目すべき有害事象(いずれかの群で0.5%以上に発現)(術後薬物療法期)

MedDRA/J v23.1、GradeはCTCAE v4.0
ー:集計なし
同一症例がカテゴリー内の事象を複数発現した場合、カテゴリー別集計(太字)は1例とカウントしている
(第4回中間解析、データカットオフ日:2021年3月23日)
安全性(併合期*)
安全性解析対象集団における副作用及び免疫関連など特に注目すべき有害事象
*術前薬物療法期及び術後薬物療法期
【安全性解析対象集団】
キイトルーダ®群:無作為化され、治療(治験薬投与又は手術)を1回以上受けたすべての患者(783例)
プラセボ群:無作為化され、治療(治験薬投与又は手術)を1回以上受けたすべての患者(389例)
■ キイトルーダ®群(783例)

■ プラセボ群(389例)

MedDRA/J v23.1、GradeはCTCAE v4.0
(第4回中間解析、データカットオフ日: 2021年3月23日)
■主な副作用(いずれかの群で発現率10%以上)
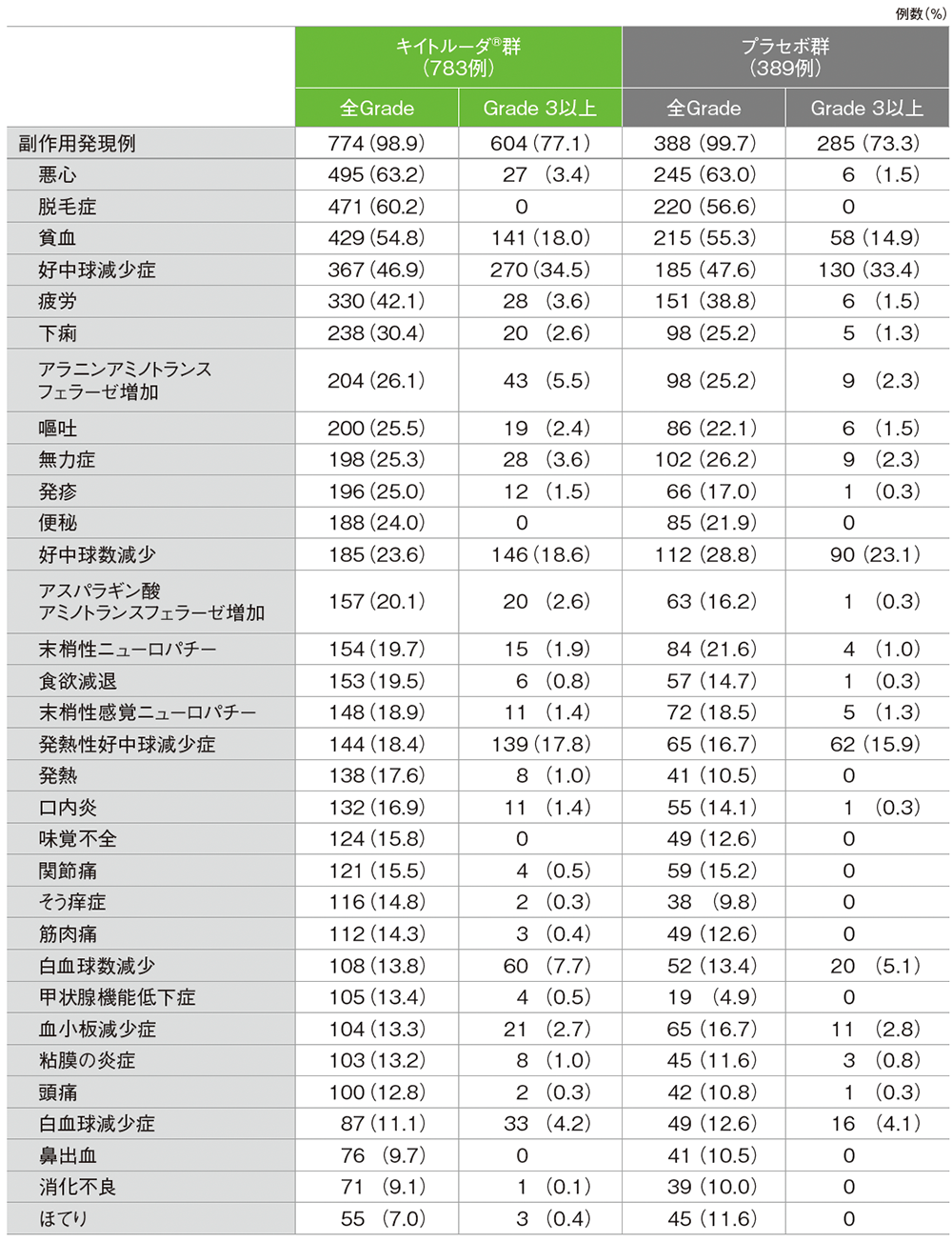
MedDRA/J v23.1、GradeはCTCAE v4.0
(第4回中間解析、データカットオフ日: 2021年3月23日)
■免疫関連など特に注目すべき有害事象(いずれかの群で発現率>0%)(併合期)

MedDRA/J v23.1、GradeはCTCAE v4.0
(第4回中間解析、データカットオフ日:2021年3月23日)
安全性解析対象集団における副作用及び免疫関連など特に注目すべき有害事象
【安全性解析対象集団】
キイトルーダ®群:無作為化され、治療(治験薬投与又は手術)を1回以上受けたすべての患者(783例)
プラセボ群:無作為化され、治療(治験薬投与又は手術)を1回以上受けたすべての患者(389例)
■ キイトルーダ®群(783例)

■ プラセボ群(389例)

(OS最終解析、データカットオフ日: 2024年3月22日、追跡期間中央値: 75.1ヵ月)
■主な副作用(いずれかの群で発現率10%以上)
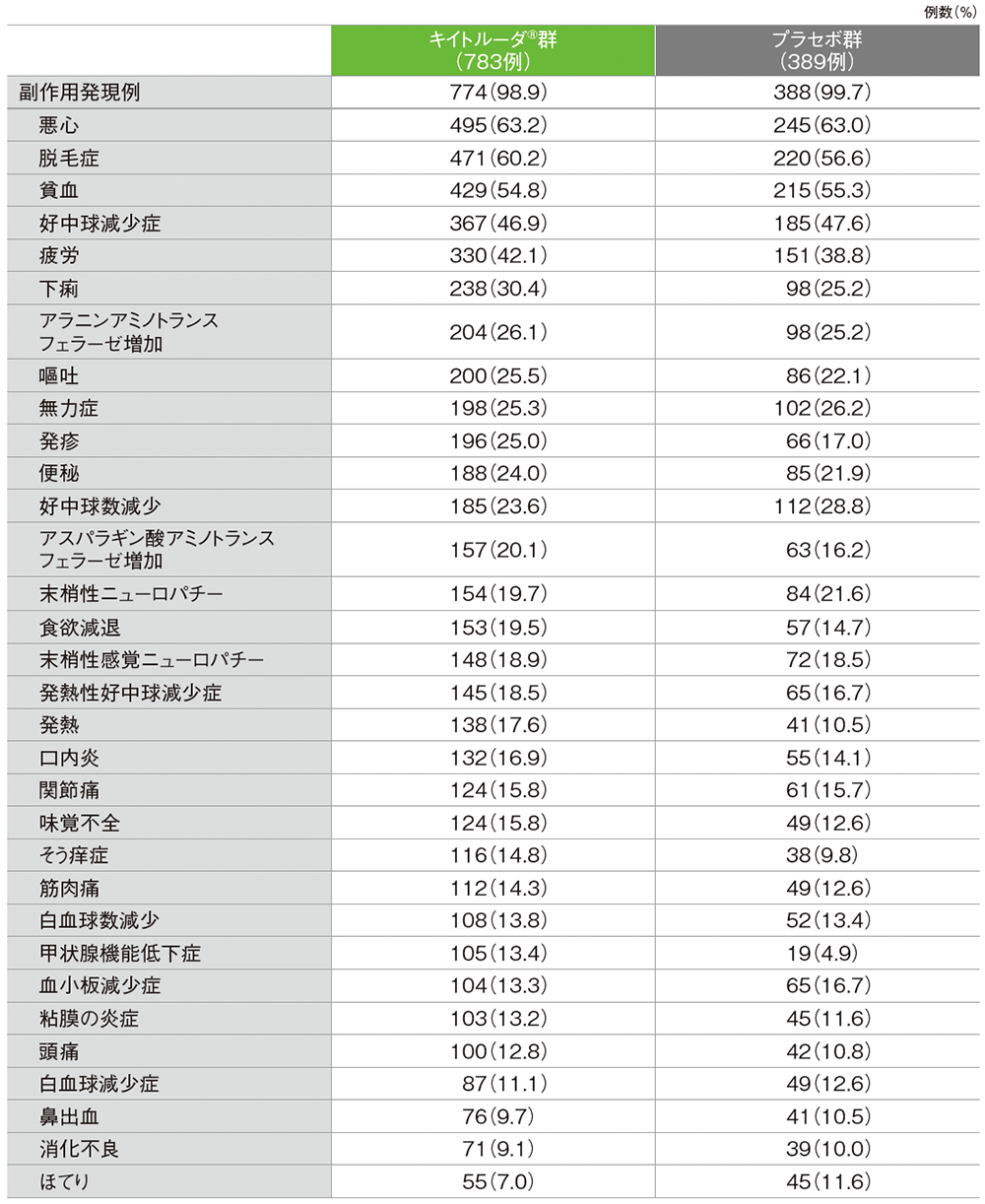
(OS最終解析、データカットオフ日: 2024年3月22日、追跡期間中央値: 75.1ヵ月)
免疫関連など特に注目すべき有害事象は、キイトルーダ®群で351/783例(44.8%)、プラセボ群で89/389例(22.9%)に認められました。
■ 免疫関連など特に注目すべき有害事象(いずれかの群で発現率>0%)
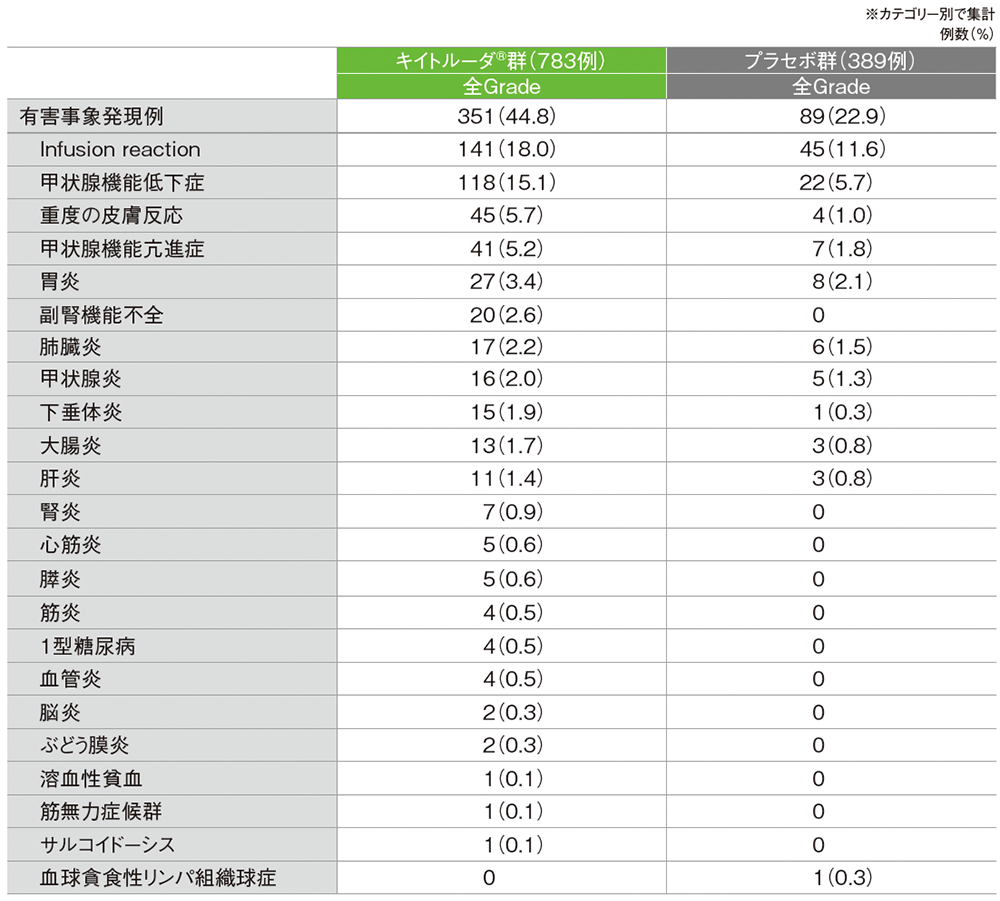
(OS最終解析、データカットオフ日: 2024年3月22日、追跡期間中央値: 75.1ヵ月)
Q&A
Q1 妊婦又は妊娠している可能性のある女性に投与してもよいですか?
A 妊婦又は妊娠している可能性のある女性には、治療上の有益性が危険性を上回ると判断した場合にのみ投与してください。
本剤を用いた生殖発生毒性試験は実施されていません。妊娠マウスに抗PD-1抗体又は抗PD-L1抗体を投与すると、流産率が増加することが報告されていることから、妊娠中の女性に対する本剤の投与は、胎児に対して有害な影響を及ぼす可能性があります1)。
Q2 授乳中の女性に投与してもよいですか?
A 授乳中の女性に投与する場合には、治療上の有益性及び母乳栄養の有益性を考慮し、授乳の継続又は中止を検討してください。
本剤のヒト母乳中への移行に関するデータはありませんが、ヒトIgGは母乳中に移行することが知られています1)。
Q3 KEYNOTE-522試験プロトコールでは、G-CSF製剤の使用に関する規定はありますか?
A 術前薬物療法①においては、各サイクルの化学療法投与後にG-CSF製剤の投与が可能でした。術前薬物療法②においては、各サイクルの化学療法投与後にG-CSF製剤又は持続型G-CSF製剤の投与が可能でした2)。
ただし、日本においてがん化学療法による発熱性好中球減少症(FN)の発症抑制に対して、G-CSF製剤(フィルグラスチム、レノグラスチム)の使用は適応外となります。
なお、KEYNOTE-522試験で実際にG-CSF製剤の予防投与が実施された割合については不明です。
G-CSF製剤の使用にあたっては、各製品電子添文を参照してください。
Q4 術前又は術後薬物療法の効果判定のタイミングは?
A KEYNOTE-522試験のプロトコールでは、術前薬物療法期の疾患進行/再発の評価*1は2サイクル目以降各サイクルの1日目、治験薬投与の前に実施していました。また、pCR評価*2のために、根治手術時に採取した腫瘍組織について病理学的腫瘍病期分類が行われました。術後薬物療法期の再発の評価*1は、各サイクルの1日目、治験薬投与の前に実施していました2)。
*1:身体所見、画像検査、生検及び/又は手術による評価
*2:全身性の術前薬物療法完了後、根治手術で完全切除された乳房標本及び採取されたすべての所属リンパ節をヘマトキシリン・エオジン染色後、治験実施医療機関の病理医が盲検下で浸潤がんの消失を評価した。
Q5 KEYNOTE-522試験において、術前薬物療法後に手術を実施できましたか?
A 手術が実施できた割合は、キイトルーダ®群98.0%、プラセボ群97.7%でした3)。
Q6 KEYNOTE-522試験において、術前薬物療法でpCRを達成した場合、術後薬物療法の省略は可能でしたか?(術前薬物療法でキイトルーダ®の有害事象による中止の場合は除く)
A 術前薬物療法によるpCRの達成有無に関わらず全患者が術後薬物療法を受ける試験デザインでした2)。pCR達成症例で術後のキイトルーダ®投与を省略したエビデンスはありませんので、試験デザイン通りキイトルーダ®単剤による術後薬物療法を行っていただくことを推奨いたします。
Q7 術前薬物療法②のAC又はEC療法はdose-dense療法でもいいですか?
A KEYNOTE-522試験のプロトコールでは、AC又はEC療法は3週間間隔で投与することと規定しており2)、dose-dense療法で投与した場合の有効性、安全性は検討されておりません。なお、キイトルーダ®電子添文の「用法及び用量に関連する注意」の項には、『本剤の用法は「17. 臨床成績」の項の内容を熟知し選択すること。また、併用する他の抗悪性腫瘍剤は「17. 臨床成績」の項の内容を熟知し、国内外の最新のガイドライン等を参考にした上で、選択すること。[17.1.27参照]』と記載しています4)。
Q8 KEYNOTE-522試験において、術後薬物療法を実施できなかった患者の割合は?
A KEYNOTE-522試験で無作為化されたすべての患者のうち、術前薬物療法期で中止し術後薬物療法を実施できなかった患者は、キイトルーダ®群で24.2%、プラセボ群で14.9%でした。主な中止理由は有害事象でした3)。
Q9 術後の放射線療法とキイトルーダ®投与のタイミングはどのように規定されていますか?
A キイトルーダ®の電子添文上、放射線療法中もしくは放射線療法後の患者さんへのキイトルーダ®投与に関する記載はありません4)。
KEYNOTE-522試験では、キイトルーダ®は放射線療法と同時又は放射線療法終了2週間後に投与を開始することとされていました2)。
Q10 KEYNOTE-522試験において、術前薬物療法を完遂出来た患者の割合は?
A 術前薬物療法①を完遂したキイトルーダ®群の患者の割合は87.9%(684例/778例)、プラセボ群で91.5%(356例/389例)でした。また、術前薬物療法②を完遂したキイトルーダ®群の患者の割合は90.9%(660例/726例)、プラセボ群で93.0%(343例/369例)でした。中止に至った理由として最も多かったものはいずれの群も有害事象でした3)。
1)キイトルーダ® 適正使用ガイド
2)Schmid P et al. N Engl J Med 2022; 386: 556-567(Protocol)
KEYNOTE-522試験はMSD社の資金提供により行われた。
3)承認時評価資料: 国際共同第Ⅲ相試験(KEYNOTE-522試験)
4)キイトルーダ® 電子添文
キイトルーダ®・悪性腫瘍関連領域情報


キイトルーダ®治療日誌:<頭頸部癌>キイトルーダ®術前補助療法
