KEYNOTE-355試験(進行再発)
本剤は、一部承認外の効能又は効果による臨床試験の成績も含めた臨床データパッケージで評価され、承認されました。そのため、国内で承認されている効能又は効果と異なるデータも紹介しています。
<進行再発トリプルネガティブ乳癌>
トリプルネガティブ乳癌:国際共同臨床試験成績:国際共同第Ⅲ相試験<KEYNOTE-355試験>
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-355試験)
社内資料:国際共同第Ⅲ相試験(KEYNOTE-355試験)安全性
Cortes J et al. Lancet 2020; 396: 1817-1828
Cortes J et al. Lancet 2020; 396: 1817-1828 Supplementary Data(Protocol)
Cortes J et al. N Engl J Med 2022; 387: 217-226
本試験はMSD社の資金提供により行われた。Javier CortesはMSD社から顧問料などを受領している。また、著者のうち、Zifang Guo、Jing Zhao、Vassiliki Karantzaは同社の社員である。その他の著者にMSD社より講演料、顧問料などを受領している者が含まれる。
試験概要
※本試験は、パート1及びパート2から構成された。
【 目的 】転移・再発乳癌に対する全身性の前治療歴のない、切除不能な転移・再発又は局所進行性のトリプルネガティブ乳癌患者におけるキイトルーダ®と化学療法の有効性及び安全性を比較検討する。
【デザイン】国際共同無作為化二重盲検第Ⅲ相試験[優越性試験]
[パート1(非盲検非無作為化Safety run-in試験)、パート2(二重盲検無作為化試験)][第2回中間解析結果(データカットオフ日: 2019年12月11日)][最終解析結果(データカットオフ日:2021年6月15日)]
【 対象 】転移・再発乳癌に対する全身性の前治療歴のない、切除不能な転移・再発又は局所進行性のトリプルネガティブ乳癌患者*1
有効性解析対象集団: 847例(うち日本人患者87例を含む)[パート2]
安全性解析対象集団†: 877例(うち日本人患者91例を含む)[パート1及びパート2]
† 最終解析の安全性解析は「パート2]のみを対象
【 方法 】
パート1 :キイトルーダ®200mgを3週間間隔で点滴静注と化学療法併用〔ゲムシタビン1000mg/m2及びカルボプラチンAUC 2に相当する量を1週間間隔で2回点滴静注(21日を1サイクル)、パクリタキセル90mg/m2を1週間間隔で3回点滴静注(28日を1サイクル)又はnab-パクリタキセル100mg/m2を1週間間隔で3回点滴静注(28日を1サイクル)〕のいずれかに割り付けた。
パート2 : キイトルーダ®と化学療法併用群〔以下、キイトルーダ®併用群: キイトルーダ®200mgを3週間間隔で点滴静注とゲムシタビン1000mg/m2及びカルボプラチンAUC 2に相当する量を1週間間隔で2回点滴静注(21日を1サイクル)、パクリタキセル90mg/m2を1週間間隔で3回点滴静注(28日を1サイクル)又はnab-パクリタキセル100mg/m2を1週間間隔で3回点滴静注(28日を1サイクル)〕又はプラセボと化学療法併用群〔以下、プラセボ併用群: プラセボとゲムシタビン1000mg/m2及びカルボプラチンAUC 2に相当する量を1週間間隔で2回点滴静注(21日を1サイクル)、パクリタキセル90mg/m2を1週間間隔で3回点滴静注(28日を1サイクル)又はnab-パクリタキセル100mg/m2を1週間間隔で3回点滴静注(28日を1サイクル)〕に2:1の割合で無作為に割り付けた*2。
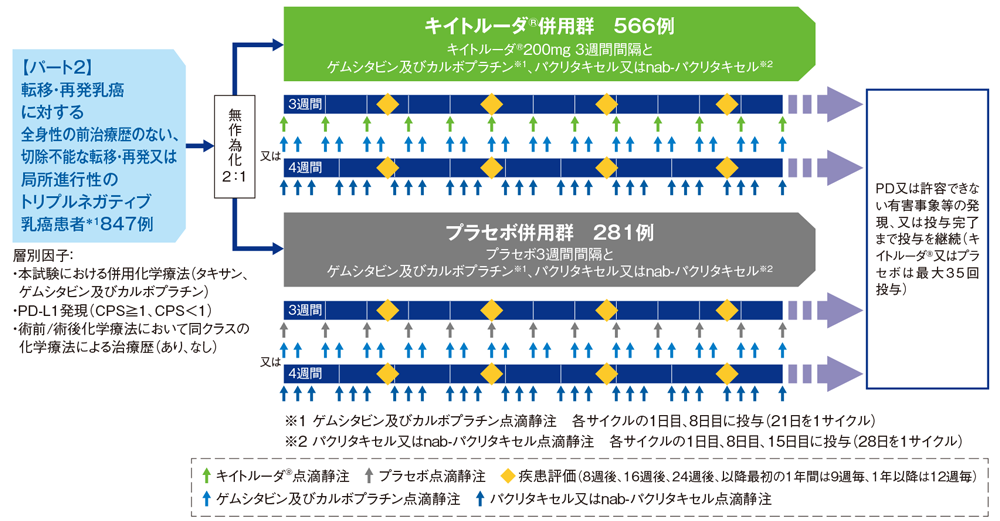
【評価項目】
主要評価項目:PD-L1発現陽性(CPS≧10及びCPS≧1)患者、全体集団における無増悪生存期間(progression free survival: PFS)※1及び全生存期間(overall survival: OS)※1(パート1:安全性及び忍容性)
副次評価項目: PD-L1発現陽性(CPS≧10及びCPS≧1)患者、全体集団における奏効率(overall response rate: ORR)※2、奏効期間(duration of response: DOR)及び病勢コントロール率(disease control rate: DCR)、安全性解析対象集団の安全性及び忍容性
※1 検証的解析項目 ※2 検証的解析項目(CPS≧10を除く)
【判定基準】
PFS、ORR、DOR及びDCRは、盲検下独立中央画像判定機関がRECISTガイドライン1.1版に基づき判定した。
【解析計画】
解析対象集団:対照群との有効性の比較はITT*3集団(パート2)、安全性はASaT*4集団(パート1及びパート2)を解析対象とした。
有効性評価の統計手法:PFS、OSはKaplan-Meier法を用いて推定した。PFS及びOSの群間比較には層別ログランク検定を用いた。ハザード比(HR)及び95%信頼区間は、投与群を共変量とし、層別Cox比例ハザードモデルを用いて算出した。また、Cox比例ハザードモデルを用いて、年齢(<65歳、≧65歳)、地域(ヨーロッパ/北アメリカ/オーストラリア、アジア、その他)、併用化学療法(ゲムシタビン及びカルボプラチン、パクリタキセル、nab-パクリタキセル)、同クラスの術前/術後化学療法歴(あり、なし)、術前/術後化学療法歴(あり、なし)、タキサンによる術前/術後化学療法歴(あり、なし)、閉経状態(閉経前、閉経後)、ECOG PS*5(0、1)、無病期間(De novo転移、<12ヵ月、≧12ヵ月)、転移臓器数(<3、≧3)等についてのPFS、OSの部分集団解析を実施した。DORはKaplan-Meier法を用いて要約した。日本人集団については、治験実施計画書に記載されていないが、PFS、ORR、DCRについても算出し、評価資料として承認時に評価された。
多重性の調整:本試験は3回の有効性の中間解析を事前に計画し、試験全体の有意水準を片側2.5%となるように厳密に制御した。PFSの解析は1回目の中間解析及び2回目の中間解析(PFSの最終解析)で実施し、OSは3回の中間解析及び最終解析を実施することとした。1回目の中間解析後に解析計画を改訂し、最初に配分する有意水準を改訂した。OSの中間解析と最終解析における有意水準の配分には群逐次検定(α消費関数)を用いた。PFS、OS及びORRの多重性の調整は、Maurer & Bretzのgraphical approachの拡張法を用いた。 PFSに関しては、PFSに配分された最初の有意水準はCPS≧10患者に割り当て、CPS≧10患者、CPS≧1患者、全体集団の順番に、これら3つのPFSにおいて階層的検定手順を用いることとした。OSに関しては、OSに配分された最初の有意水準はCPS≧10患者とCPS≧1患者に分割した。CPS≧10患者のPFS、全体集団のPFSが統計学的に有意であった場合、それらの有意水準(CPS≧10患者については有意水準の73%)をCPS≧10患者のOSに再配分し、最初に配分した有意水準と合わせて検定することとした。CPS≧10患者のOSが統計学的に有意であった場合、CPS≧10患者のOSの有意水準をCPS≧1患者のOSに再配分し、最初に配分した有意水準と合わせて検定することとした。CPS≧1患者のOSが統計学的に有意であった場合、CPS≧1患者のOSの有意水準を全体集団のOSに再配分することとした。
*1 再発乳癌の場合は、必要なステージⅠ~Ⅲの治療を完了し、治癒目的の治療終了注)から6ヵ月以上経過していること
*2 術前/術後化学療法と同クラスの化学療法の薬剤は、治癒目的の治療終了注)から12ヵ月以上経過後の局所又は遠隔の再発の場合に使用可能であった
注)原発乳癌の切除日又は直近の術後化学療法の最終投与日のいずれか遅い日
*3 ITT(intention-to-treat)集団:無作為化されたすべての患者
*4 ASaT(all subjects as treated)集団:治験薬を1回以上投与されたすべての患者
*5 ECOG:Eastern Cooperative Oncology Group PS:performance status
4. 効能又は効果(抜粋)
〇PD-L1陽性のホルモン受容体陰性かつHER2陰性の手術不能又は再発乳癌
5. 効能又は効果に関連する注意(抜粋)
〈PD-L1陽性のホルモン受容体陰性かつHER2陰性の手術不能又は再発乳癌〉
5.20 PD-L1発現率(CPS)について、「17. 臨床成績」の項の内容を熟知し、十分な経験を有する病理医又は検査施設における検査により、PD-L1の発現が確認された患者に投与すること。検査にあたっては、承認された体外診断用医薬品又は医療機器を用いること。なお、承認された体外診断用医薬品又は医療機器に関する情報については、以下のウェブサイトから入手可能である:
https://www.pmda.go.jp/review-services/drug-reviews/review-information/cd/0001.html
[17.1.26参照]
ゲムシタビンの効能又は効果、用法及び用量は以下のとおりです。
【効能又は効果】(抜粋)手術不能又は再発乳癌
〈効能又は効果に関連する使用上の注意〉(抜粋)手術不能又は再発乳癌
本剤の術前・術後補助化学療法における有効性及安全性は確立していない。
【用法及び用量】(抜粋)
〈手術不能又は再発乳癌〉
通常、成人にはゲムシタビンとして1回1250mg/m2を30分かけて点滴静注し、週1回投与を2週連続し、3週目は休薬する。これを1コースとして投与を繰り返す。なお、患者の状態により適宜減量する。
パクリタキセルの効能又は効果、用法及び用量は以下のとおりです。
【効能又は効果】(抜粋)乳癌
【用法及び用量】(抜粋)
乳癌にはA法又はB法を使用する。
A法:通常、成人にはパクリタキセルとして、1日1回210mg/m2(体表面積)を3時間かけて点滴静注し、少なくとも3週間休薬する。これを1クールとして、投与を繰り返す。
B法:通常、成人にはパクリタキセルとして、1日1回100mg/m2(体表面積)を1時間かけて点滴静注し、週1回投与を6週連続し、少なくとも2週間休薬する。これを1クールとして、投与を繰り返す。
なお、投与量は、患者の状態により適宜減量する。
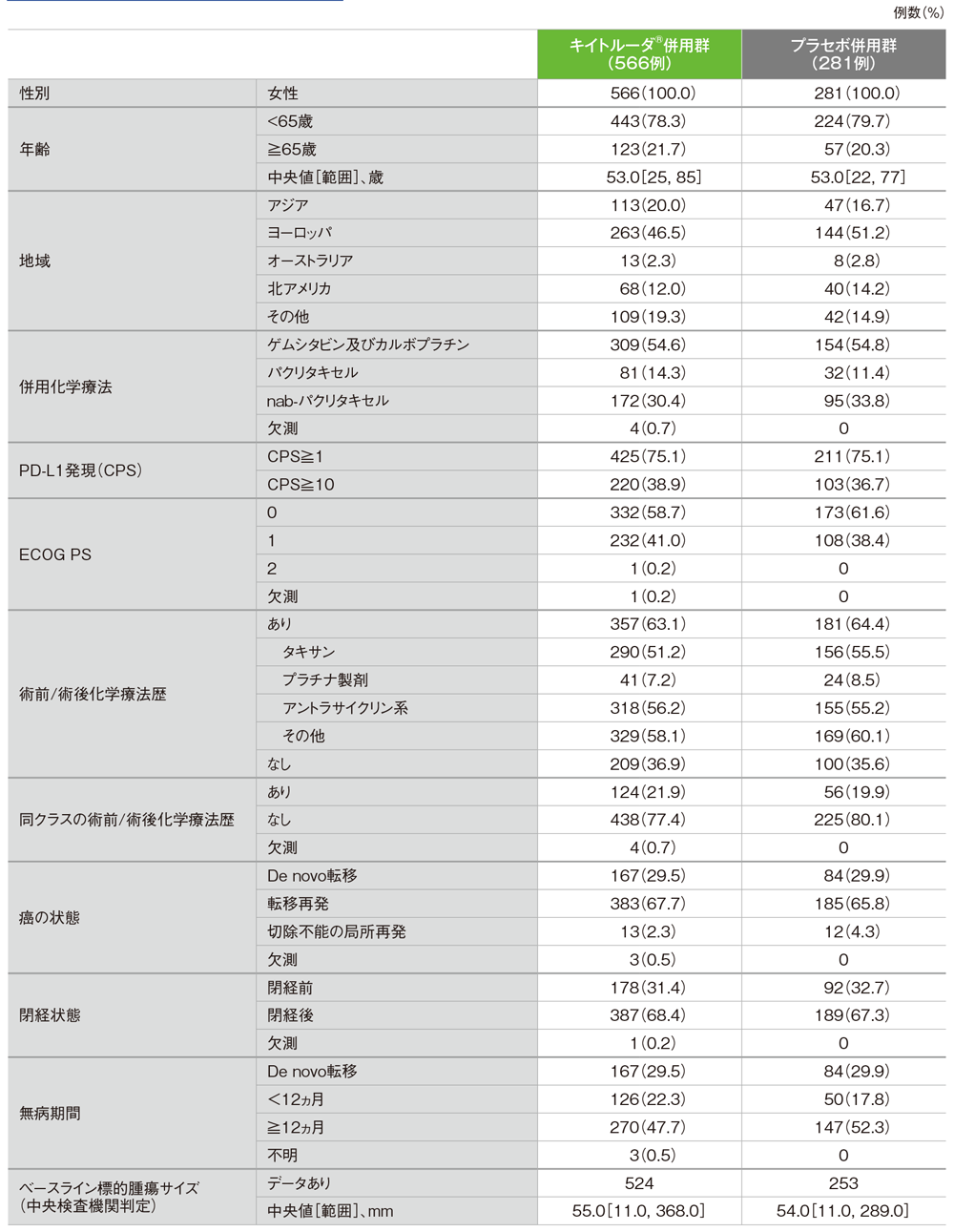
患者背景(パート2)(ITT集団)

主要評価項目:優越性試験(検証的解析結果)
PD-L1発現陽性(CPS≧10)患者における無増悪生存期間: PFS
- PFS中央値は、キイトルーダ®併用群で9.7ヵ月(95%CI: 7.6, 11.3)、プラセボ併用群で5.6ヵ月(95%CI: 5.3, 7.5)でした。プラセボ併用群に対するキイトルーダ®併用群のハザード比は、0.65(95%CI: 0.49, 0.86)で、有意にPFSを改善しました(p=0.0012、層別ログランク検定[片側]、有意水準α=0.00411、検証的解析結果)。
■PD-L1発現陽性(CPS≧10)患者における無増悪生存期間(PFS)のKaplan-Meier曲線
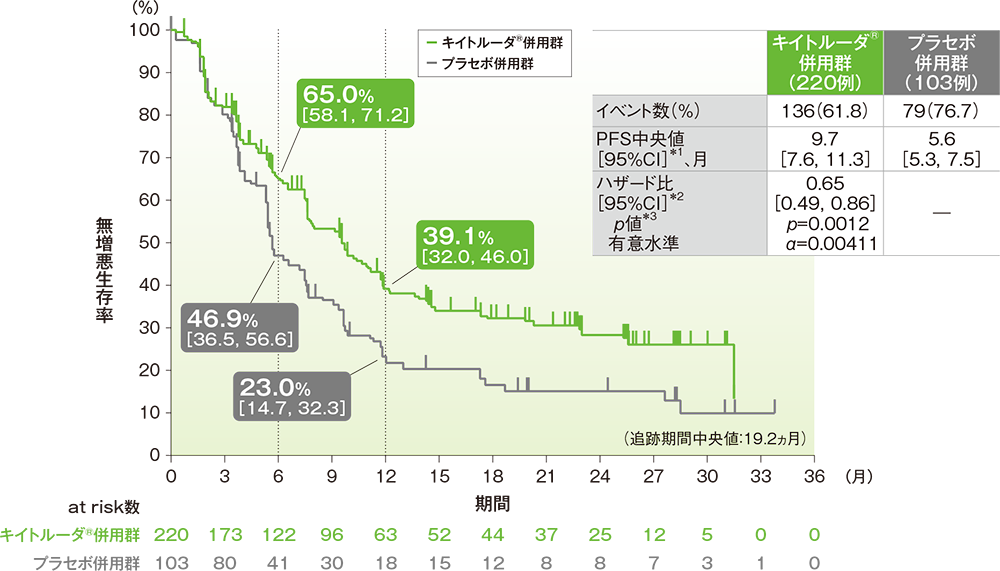
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量とし、無作為化に用いた層別因子[本試験における併用化学療法(タキサン、ゲムシタビン及びカルボプラチン)、PD-L1発現(CPS≧1、CPS<1)、術前/術後補助療法において同クラスの化学療法による治療歴(あり、なし)]を層別因子とした層別Cox比例ハザードモデルに基づく
*3 無作為化に用いた層別因子を層別因子とした層別ログランク検定[片側]に基づく
(データカットオフ日:2019年12月11日)
主要評価項目:優越性試験
PD-L1発現陽性(CPS≧1)患者及び全体集団における無増悪生存期間: PFS
- CPS≧1患者におけるプラセボ併用群に対するキイトルーダ®併用群のハザード比は0.74であり、優越性は検証されませんでした(p=0.0014、層別ログランク検定[片側]、有意水準α=0.00111、検証的解析結果)*4。全体集団におけるプラセボ併用群に対するキイトルーダ®併用群のハザード比は0.82でした(名目上のp=0.0112)。
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量とし、無作為化に用いた層別因子[本試験における併用化学療法(タキサン、ゲムシタビン及びカルボプラチン)、PD-L1発現(CPS≧1、CPS<1)、術前/術後化学療法において同クラスの化学療法による治療歴(あり、なし)]を層別因子とした層別Cox比例ハザードモデルに基づく
*3 無作為化に用いた層別因子を層別因子とした層別ログランク検定[片側]に基づく
*4 PD-L1発現陽性(CPS≧1)患者で有意差が認められなかったため検定を終了した
(データカットオフ日:2019年12月11日)
サブグループ解析 部分集団因子別の無増悪生存期間(PFS)
■PD-L1発現陽性(CPS≧10)患者における無増悪生存期間(PFS)のハザード比のフォレストプロット
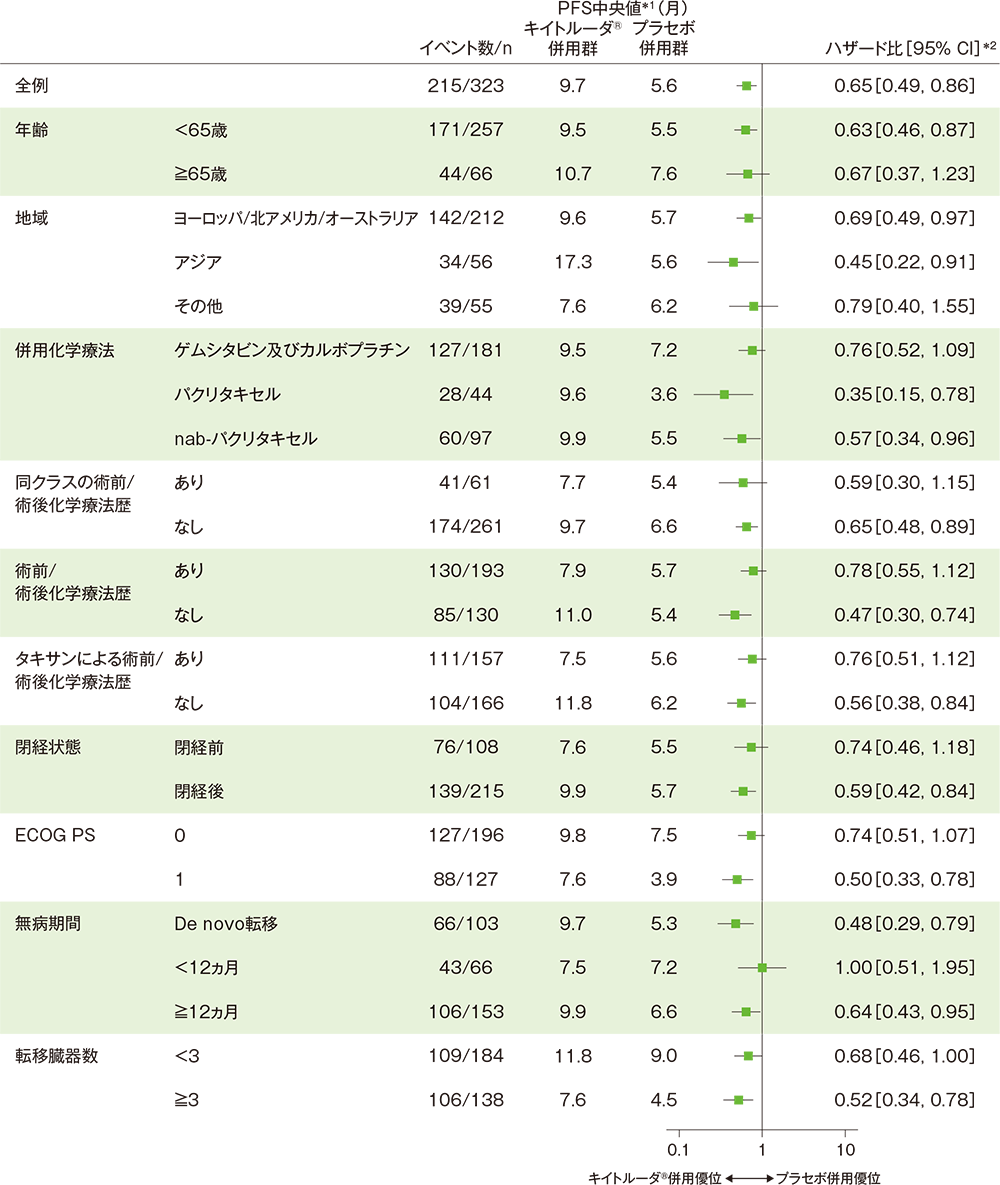
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 全例:投与群を共変量とし、無作為化に用いた層別因子[本試験における併用化学療法(タキサン、ゲムシタビン及びカルボプラチン)、PD-L1発現(CPS≧1、CPS<1)、術前/術後化学療法において同クラスの化学療法による治療歴(あり、なし)]を層別因子とした層別Cox比例ハザードモデルに基づく
サブグループ:Cox比例ハザードモデルに基づく
(追跡期間中央値:19.2ヵ月、データカットオフ日: 2019年12月11日)

主要評価項目:優越性試験(検証的解析結果)
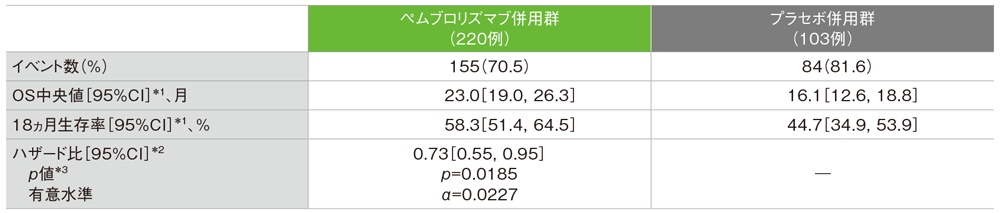
PD-L1発現陽性(CPS≧10)患者における全生存期間:OS
- OS中央値は、ペムブロリズマブ併用群で23.0ヵ月(95%CI:19.0, 26.3)、プラセボ併用群で16.1ヵ月(95%CI:12.6, 18.8)でした*1。プラセボ併用群に対するペムブロリズマブ併用群のハザード比は、0.73(95%CI:0.55, 0.95)で*2、有意にOSを改善しました(p=0.0185、層別ログランク検定[両側]、有意水準α=0.0227、検証的解析結果)*3。
■PD-L1発現陽性(CPS≧10)患者における全生存期間(OS)のKaplan-Meier曲線
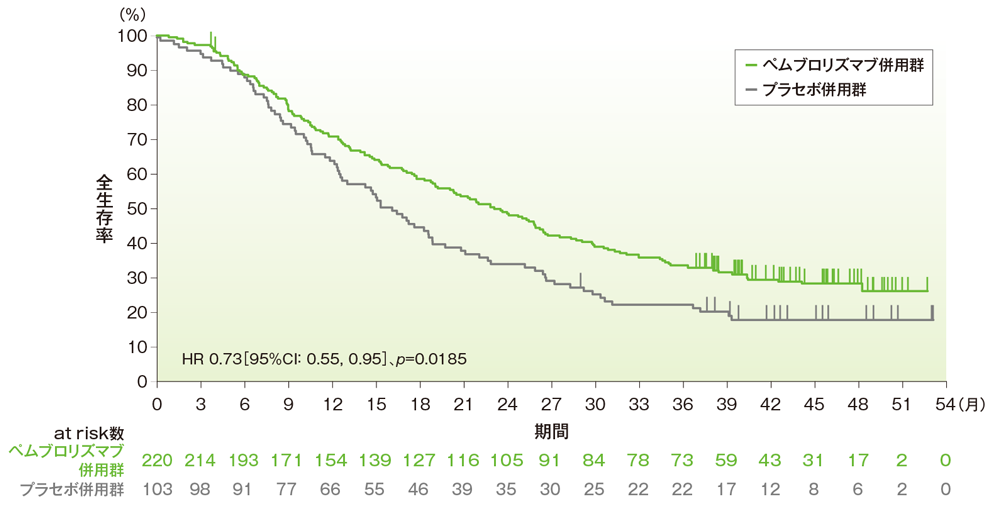
Cortes J et al. N Engl J Med 2022; 387: 217-226
Copyright ©2022 Massachusetts Medical Society. All rights reserved. Translated with permission.
本試験はMSD社の資金提供により行われた。
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量とし、無作為化に用いた層別因子[本試験における併用化学療法(タキサン、ゲムシタビン及びカルボプラチン)PD-L1発現(CPS≧1、CPS<1)、術前/術後化学療法において同クラスの化学療法による治療歴(あり、なし)]を層別因子とした層別Cox比例ハザードモデルに基づく
*3 無作為化に用いた層別因子を層別因子とした層別ログランク検定[両側]。解析計画では片側検定を行うこととしていたが、掲載誌の規定により両側検定の結果と有意水準を記載した。
(データカットオフ日:2021年6月15日)
主要評価項目:優越性試験
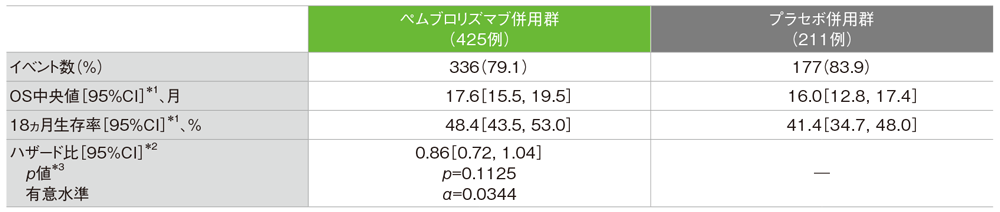
PD-L1発現陽性(CPS≧1)患者及び全体集団における全生存期間:OS
- CPS≧1患者におけるOS中央値は、ペムブロリズマブ併用群で17.6ヵ月(95%CI:15.5, 19.5)、プラセボ併用群で16.0ヵ月(95%CI:12.8, 17.4)でした*1。プラセボ併用群に対するペムブロリズマブ併用群のハザード比は0.86であり*2、優越性は検証されませんでした(p=0.1125、層別ログランク検定[両側]、有意水準α=0.0344、検証的解析結果)*3、*4。
- また全体集団におけるOS中央値は、ペムブロリズマブ併用群で17.2ヵ月(95%CI:15.3, 19.0)、プラセボ併用群で15.5ヵ月(95%CI:13.9, 17.2)でした*1。プラセボ併用群に対するペムブロリズマブ併用群のハザード比は0.89(95%CI:0.76, 1.05)でした*2。
■PD-L1発現陽性(CPS≧1)患者
Cortes J et al. N Engl J Med 2022; 387: 217-226
Copyright ©2022 Massachusetts Medical Society. All rights reserved. Translated with permission.
本試験はMSD社の資金提供により行われた。
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量とし、無作為化に用いた層別因子[本試験における併用化学療法(タキサン、ゲムシタビン及びカルボプラチン)、PD-L1発現(CPS≧1、CPS<1)、術前/術後化学療法において同クラスの化学療法による治療歴(あり、なし)]を層別因子とした層別Cox比例ハザードモデルに基づく
*3 無作為化に用いた層別因子を層別因子とした層別ログランク検定[両側]。解析計画では片側検定を行うこととしていたが、掲載誌の規定により両側検定の結果と有意水準を記載した。
*4 PD-L1発現陽性(CPS≧1)患者で有意差が認められなかったため検定を終了した
(データカットオフ日:2021年6月15日)
副次評価項目
PD-L1発現陽性(CPS≧10)患者における奏効率: ORR及び病勢コントロール率(DCR)
- ORRは、キイトルーダ®併用群53.2%(95%CI: 46.4, 59.9)、プラセボ併用群39.8%(95%CI: 30.3, 49.9)でした。
- DCR(CR、PR又は24週間以上のSDと判定した患者の割合)は、キイトルーダ®併用群65.0%(95%CI:58.3, 71.3)、プラセボ併用群54.4%(95%CI:44.3, 64.2)でした。
■PD-L1発現陽性(CPS≧10)患者における奏効率(ORR)及び病勢コントロール率(DCR)
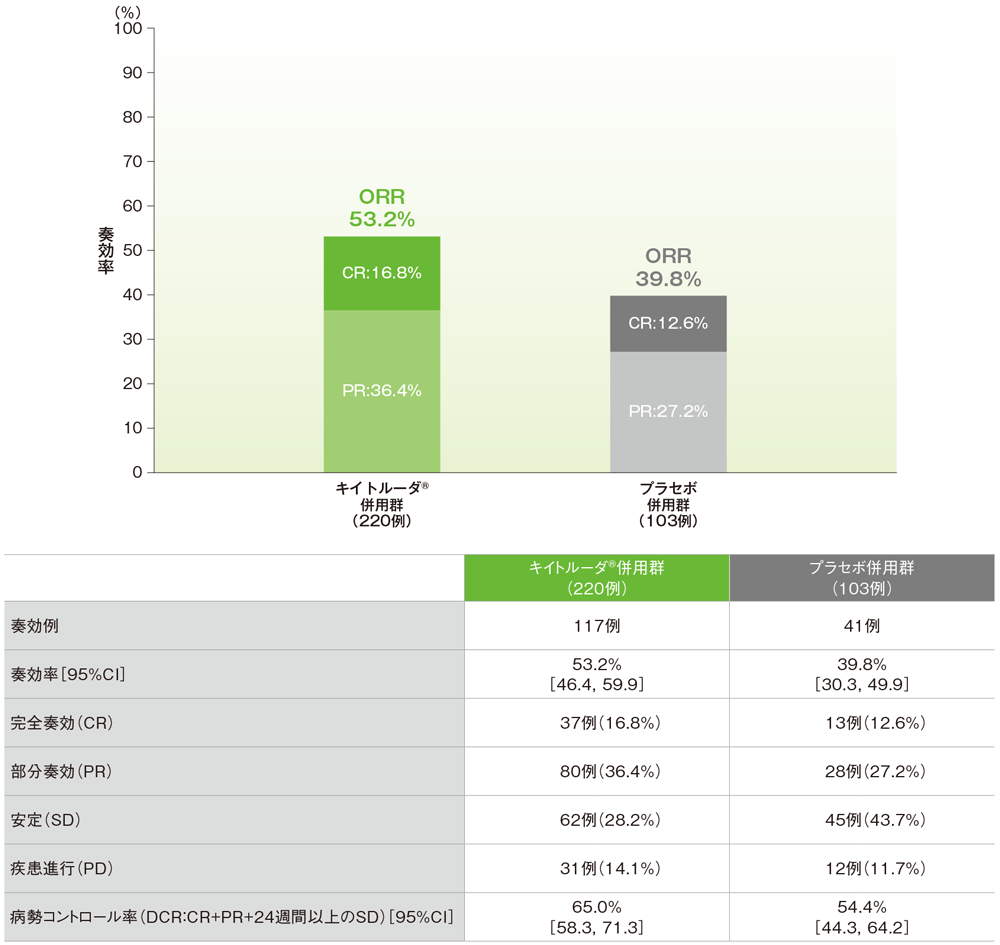
(追跡期間中央値:19.2ヵ月、データカットオフ日: 2019年12月11日)
副次評価項目
PD-L1発現陽性(CPS≧10)患者における奏効期間: DOR
- キイトルーダ®併用群において奏効が認められた117例における奏効までの期間中央値は1.9ヵ月(範囲:1.2, 11.7)、DOR中央値は19.3ヵ月(範囲:1.6+, 29.8)でした。
- プラセボ併用群において奏効が認められた41例における奏効までの期間中央値は1.9ヵ月(範囲:1.3, 7.9)、DOR中央値は7.3ヵ月(範囲:1.5, 32.5+)でした。
■PD-L1発現陽性(CPS≧10)患者における奏効期間(DOR)のKaplan-Meier曲線
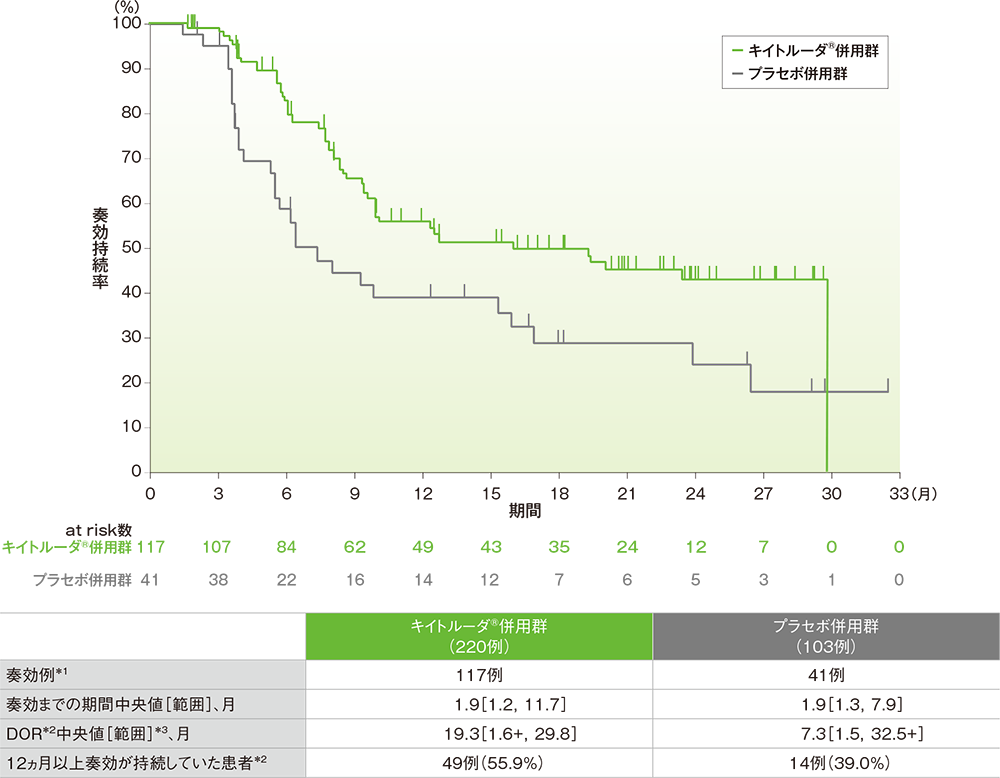
*1 完全奏効又は部分奏効を認めた症例数
*2 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*3 「+」は最後の疾患評価までPDがみられないことを示す
(追跡期間中央値:19.2ヵ月、データカットオフ日: 2019年12月11日)
安全性(パート1:主要評価項目、パート2:副次評価項目)
安全性解析対象集団の主な副作用(パート1及びパート2)
■安全性解析対象集団
キイトルーダ®併用群:パート1又はパート2のいずれかでキイトルーダ® +化学療法(ゲムシタビン及びカルボプラチン、パクリタキセル又はnab-パクリタキセル)を投与された患者(596例)
プラセボ併用群:パート2でプラセボ+化学療法(ゲムシタビン及びカルボプラチン、パクリタキセル又はnab-パクリタキセル)を投与された患者(281例)
■キイトルーダ®併用群
- 副作用:574/596例(96.3%)主な副作用(発現率20%以上):貧血291例(48.8%)、好中球減少症241例(40.4%)、悪心229例(38.4%)、脱毛症197例(33.1%)、疲労164例(27.5%)、好中球数減少132例(22.1%)
- 重篤な副作用:105/596例(17.6%)重篤な副作用(1%以上):貧血13例(2.2%)、血小板減少症10例(1.7%)、発熱性好中球減少症及び嘔吐各8例(1.3%)、肺臓炎7例(1.2%)、発熱6例(1.0%)
- 副作用によるいずれかの薬剤の中止:111例(18.6%)副作用による中止(1%以上):アラニンアミノトランスフェラーゼ増加12例(2.0%)、好中球減少症10例(1.7%)、アスパラギン酸アミノトランスフェラーゼ増加9例(1.5%)、末梢性ニューロパチー8例(1.3%)、好中球数減少及び肺臓炎各7例(1.2%)、末梢性感覚ニューロパチー6例(1.0%)
- 副作用による死亡:2例(急性腎障害及び肺炎各1例)
■プラセボ併用群
- 副作用:267/281例(95.0%)主な副作用(発現率20%以上):貧血129例(45.9%)、悪心115例(40.9%)、好中球減少症107例(38.1%)、脱毛症94例(33.5%)、疲労83例(29.5%)、好中球数減少74例(26.3%)
- 重篤な副作用:34/281例(12.1%)重篤な副作用(1%以上):貧血及び好中球減少症各4例(1.4%)、血小板減少症、発熱性好中球減少症、嘔吐及び発熱各3例(1.1%)
- 副作用によるいずれかの薬剤の中止:31例(11.0%)副作用による中止(1%以上):血小板減少症及びアラニンアミノトランスフェラーゼ増加各4例(1.4%)、好中球数減少及び末梢性ニューロパチー各3例(1.1%)
- 副作用による死亡は認められなかった
■主な副作用(いずれかの群で発現率5%以上)
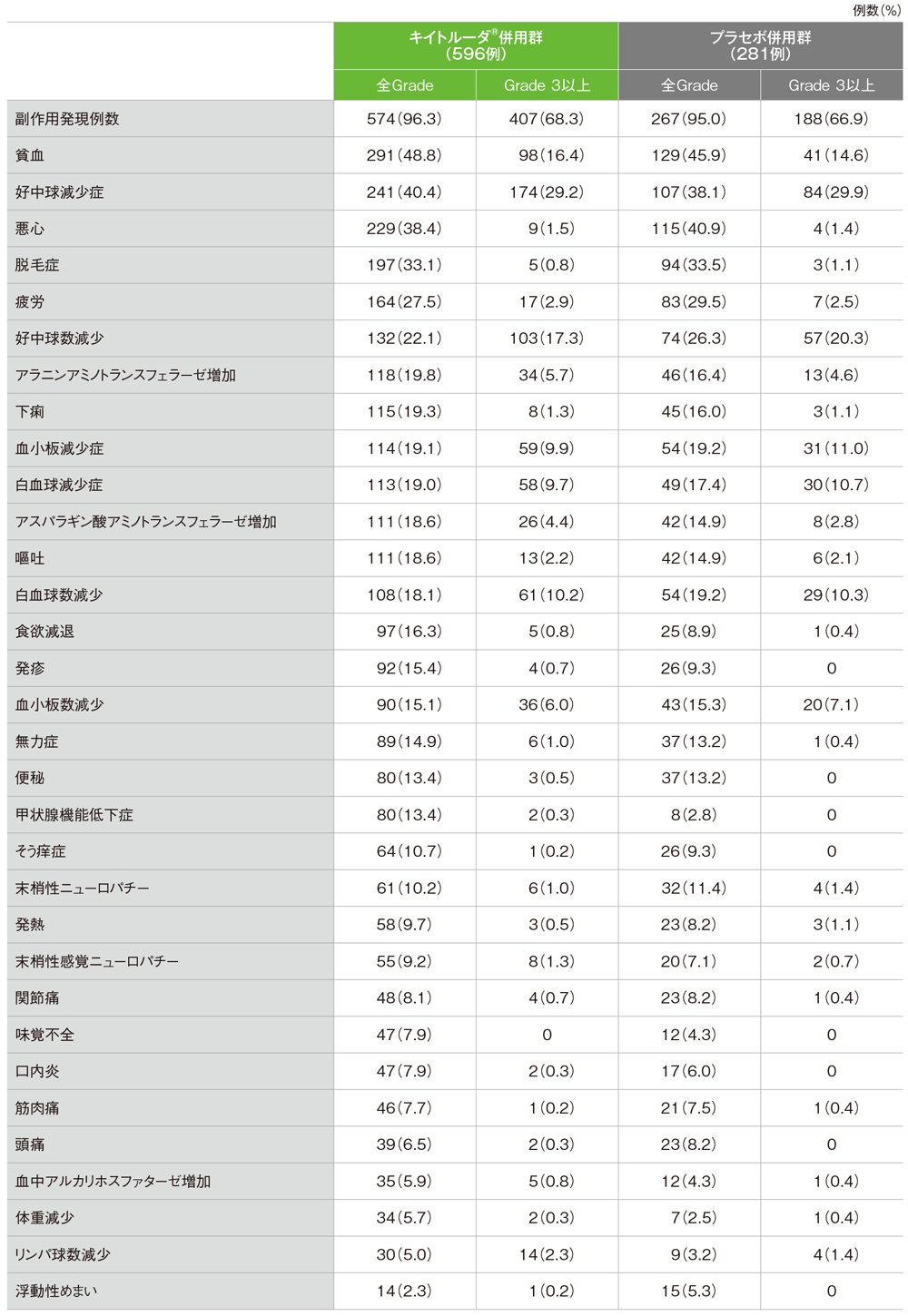
MedDRA/J version 22.1、GradeはCTCAE version 4.03
安全性:パート1-主要評価項目 パート2-副次評価項目
安全性解析対象集団の免疫関連など特に注目すべき有害事象(パート1及びパート2)
免疫関連などの有害事象は、キイトルーダ®併用群で168/596例(28.2%)、プラセボ併用群で30/281例(10.7%)に認められました。
■免疫関連など特に注目すべき有害事象(いずれかの群で>0%)
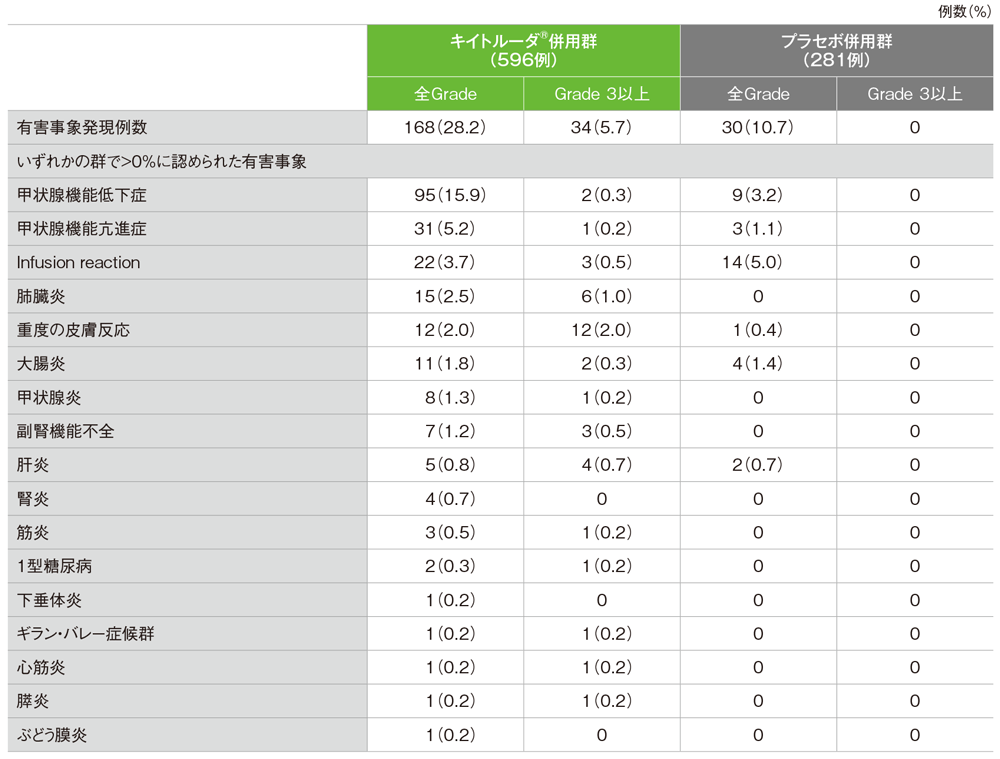
MedDRA/J version 22.1、GradeはCTCAE version 4.03
安全性:副次評価項目
安全性解析対象集団の主な副作用(パート2)
■安全性解析対象集団
- キイトルーダ®併用群:キイトルーダ®+化学療法(ゲムシタビン及びカルボプラチン、パクリタキセル又はnab-パクリタキセル)を投与された患者(562例)
- プラセボ併用群:プラセボ+化学療法(ゲムシタビン及びカルボプラチン、パクリタキセル又はnab-パクリタキセル)を投与された患者(281例)
■キイトルーダ®併用群
- 副作用:541/562例(96.3%) 主な副作用(発現率20%以上):貧血276例(49.1%)、好中球減少症231例(41.1%)、悪心221例(39.3%)、脱毛症186例(33.1%)、疲労161例(28.6%)、好中球数減少126例(22.4%)、アラニンアミノトランスフェラーゼ増加115例(20.5%)
- 重篤な副作用:100/562例(17.8%) 重篤な副作用(発現率1%以上):貧血11例(2.0%)、血小板減少症9例(1.6%)、嘔吐8例(1.4%)、発熱性好中球減少症、肺臓炎及び発熱各6例(1.1%)
- 副作用によるキイトルーダ®の中止:51例/562例(9.1%) 副作用による中止(発現率1%以上):アラニンアミノトランスフェラーゼ増加12例(2.1%)、アスパラギン酸アミノトランスフェラーゼ増加9例(1.6%)、肺臓炎7例(1.2%)
- 副作用による化学療法の中止:75/562例(13.3%) 副作用による中止(発現率1%以上):好中球減少症10例(1.8%)、末梢性ニューロパチー7例(1.2%)、好中球数減少及び末梢性感覚ニューロパチー各6例(1.1%)
- 副作用による死亡:2例(急性腎障害及び肺炎各1例)
■プラセボ併用群
- 副作用:267/281例(95.0%) 主な副作用(発現率20%以上):貧血129例(45.9%)、悪心116例(41.3%)、好中球減少症107例(38.1%)、脱毛症94例(33.5%)、疲労84例(29.9%)、好中球数減少74例(26.3%)
- 重篤な副作用:34/281例(12.1%) 重篤な副作用(発現率1%以上):貧血及び好中球減少症各4例(1.4%)、血小板減少症、嘔吐、発熱性好中球減少症及び発熱各3例(1.1%)
- 副作用によるプラセボの中止:9例/281例(3.2%) 副作用による中止(発現率1%以上):アラニンアミノトランスフェラーゼ増加4例(1.4%)
- 副作用による化学療法の中止:26/281例(9.3%) 副作用による中止(発現率1%以上):血小板減少症4例(1.4%)、末梢性ニューロパチー及び好中球数減少各3例(1.1%)
- 副作用による死亡は認められなかった
■主な副作用(いずれかの群で発現率5%以上)
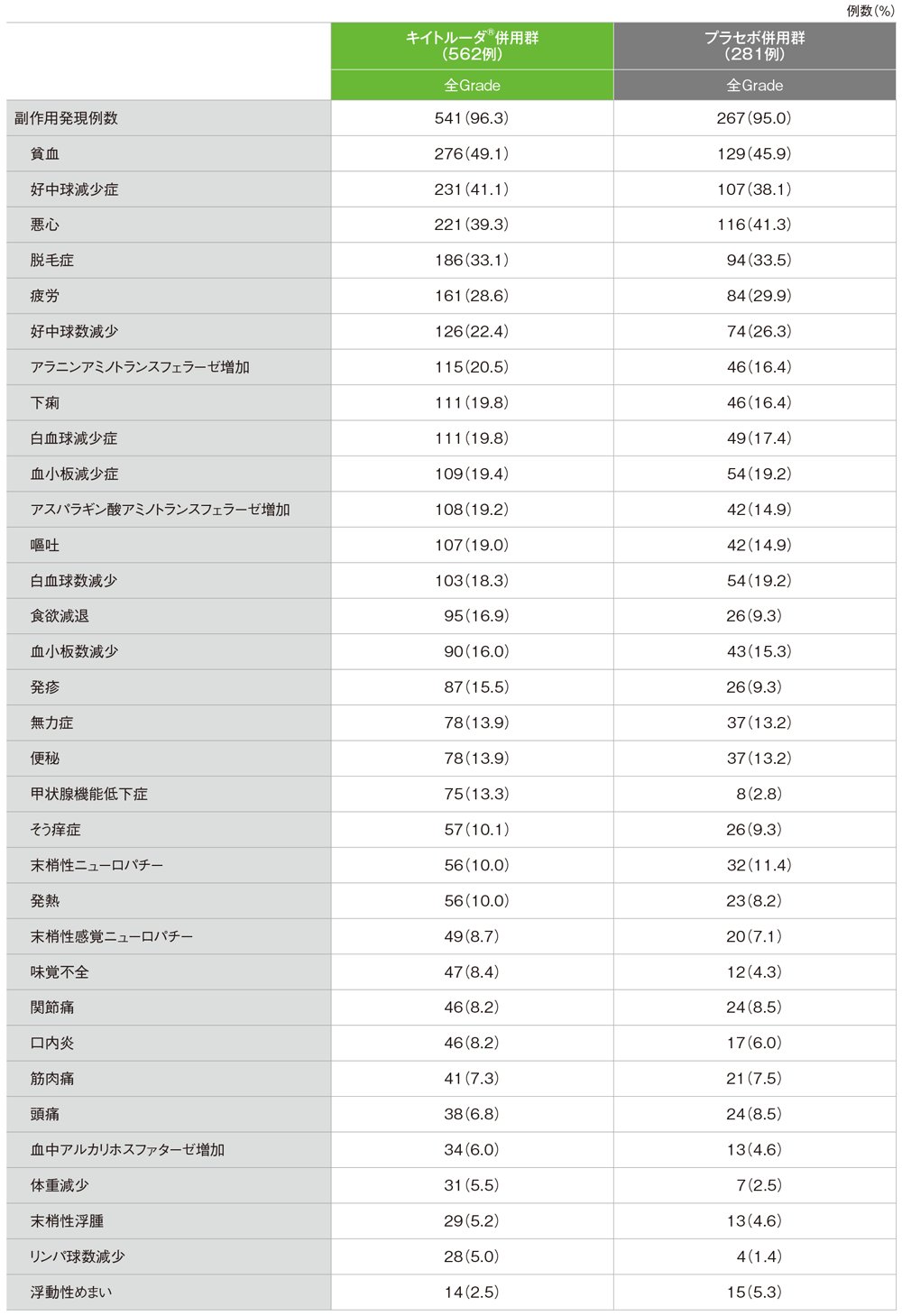
MedDRA/J version 22.1、GradeはCTCAE version 4.03
キイトルーダ®・悪性腫瘍関連領域情報


キイトルーダ®治療日誌:<頭頸部癌>キイトルーダ®術前補助療法
