成人<臓器移植> 第Ⅲ相国内試験(042試験)
臨床成績
成人<臓器移植>
第Ⅲ相国内試験(042試験)1)
日本人成人腎移植患者を対象にCMV感染及び感染症の予防*を目的としてプレバイミス®を投与した際の、安全性、有効性及び薬物動態を評価する単群非盲検第Ⅲ相試験
1)適応追加(臓器移植)承認時評価資料(第Ⅲ相国内試験:042試験)
*:プレバイミス®の効能又は効果は、「下記におけるサイトメガロウイルス感染症の発症抑制 〇同種造血幹細胞移植 〇臓器移植」である。
試験概要
主要目的:
有害事象を発現した患者の割合に基づき、プレバイミス®の安全性及び忍容性を評価する。
対象:
CMV抗体陽性ドナー/CMV抗体陰性レシピエント(D+/R−)、又はCMV抗体陽性レシピエント(R+)[ドナーのCMV抗体は陽性、陰性いずれも許容した(D+/R+又はD−/R+)]のいずれかの同種腎移植を受け、割付け時点で腎移植後0日(移植当日)から7日以内の成人患者22例(組み入れ症例)
試験デザイン:
国内多施設共同単群非盲検第Ⅲ相試験
投与方法:
移植後7日以内にプレバイミス®480mg(シクロスポリン併用時には240mg)を1日1回経口投与し、28週まで継続した。嚥下不能及び/又は錠剤の吸収を妨げる可能性がある状態の患者には、同じ用法及び用量で静脈内投与した。投与完了後、52週まで追跡して遅発性のCMV感染症を評価した。
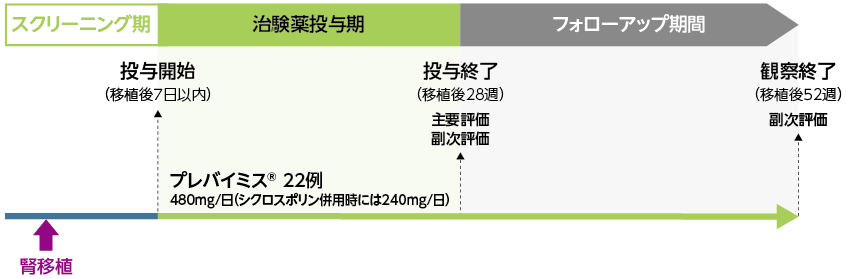
評価項目:
【主要評価項目】 有害事象及び治験薬の投与中止に至った有害事象※1を発現した患者の割合
【副次評価項目】 移植後28週及び52週以内の中央判定によるCMV感染症※2を発症した患者の割合
移植後28週及び52週以内の中央判定によるCMV感染症※2を発症又は抗CMV療法を開始※3した患者の割合
移植後28週及び52週以内の定量可能なCMV DNA血症(中央検査機関で測定)がみられた患者の割合
【探索的評価項目】 移植後52週以内のCMVの耐性変異 等
※1:有害事象の評価期間は、割付け以降治験薬最終投与後14日まで収集した(治験薬投与期)。また、重篤な副作用については、治験薬投与期として規定した期間外であっても収集することとした。
※2:CMV感染症は、「臓器障害を伴うCMV感染症」及び「CMV症候群」の2種類を含むものとした。
CMV症候群は、血中でCMVが確認され(ウイルス分離/迅速培養/抗原血症法/核酸検査)、以下の基準のうち2項目以上に該当するものとした;①2日以上継続する38℃以上の発熱、②倦怠感あるいは疲労の新規発症又は増悪、③24時間以上の間隔で2回測定した結果に基づく白血球減少症又は好中球減少症、④異型リンパ球が5%以上、⑤血小板減少症、⑥ALT又はASTが基準値上限の2倍以上
※3:抗CMV療法の開始は以下のいずれかに基づく:①CMV抗原血症法陽性(治験実施医療機関で測定)、②定量可能なCMV DNA血症(治験実施医療機関で測定)
【薬物動態】 日本人腎移植患者におけるプレバイミス®の薬物動態の評価
【安全性】 有害事象及び治験薬の投与中止に至った有害事象※1を発現した患者の割合
解析計画:
- 有効性の解析対象集団は、治験薬の投与を1回以上受け、D+/R−、D+/R+又はD−/R+であり、投与開始1日目にCMV DNAが検出されない(中央検査機関にて測定)すべての患者から構成される最大の解析対象集団[FAS(Full analysis set)]とした。
- 安全性の解析対象集団は、治験薬の投与を1回以上受けたすべての患者から構成されるAPaT(All Participants as Treated)集団とした。
- それぞれの有効性評価項目について、患者の割合及び95%信頼区間(CI)を評価時点ごとに算出した。CIは、Clopper and Pearson法による正確二項検定を用いて算出した。
- ドナー及びレシピエントのCMV抗体保有状況による結果を評価するため、D+/R−及びR+(D+/R+又はD−/R+)の部分集団別に有効性評価項目を要約した。
- 欠測値(非完了例)は非無効例として扱った[OF(Observed Failure)アプローチ]。欠測値(非完了例)を無効例とする感度分析も実施した[NC=F(Non-Completer=Failure)アプローチ]。
- 試験中止/試験完了例はその時点で打切りとした。
- 安全性は、有害事象を発現した患者数及び発現割合を要約した。また、臨床検査値、バイタルサイン及び心電図の変動を要約した。
解析対象例数:
有効性解析対象例数(FAS):21例
安全性解析対象例数(APaT):22例
(注射剤の投与を1回以上受けた患者はいなかった)
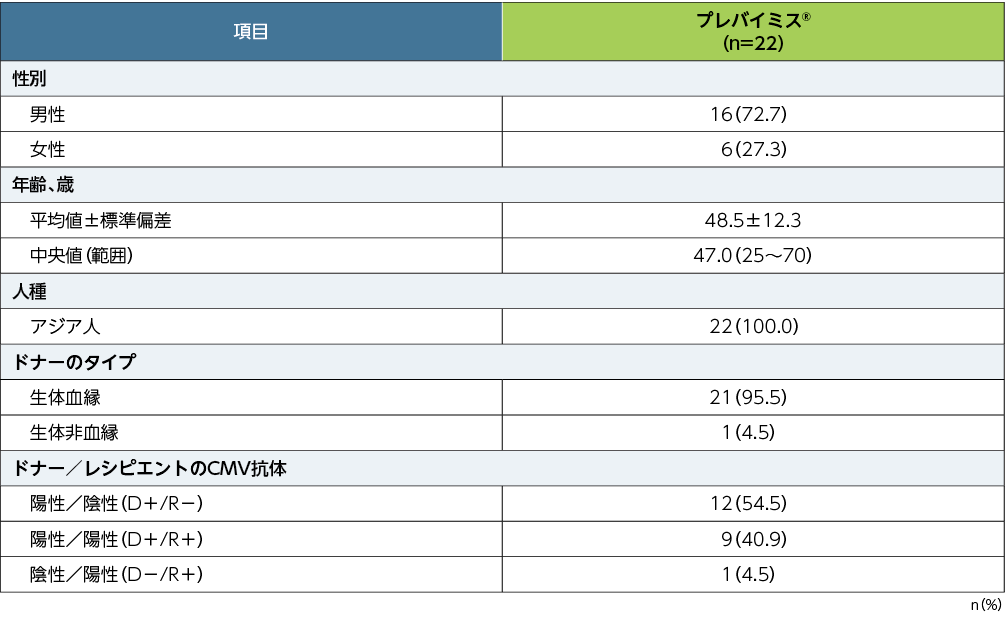
患者背景
■患者背景(APaT)
1. 有害事象及び治験薬の投与中止に至った有害事象を発現した患者の割合(主要評価項目)
本試験における有害事象の発現割合は90.9%で、プレバイミス®の投与中止に至った有害事象の発現割合は13.6%でした。
■有害事象及び治験薬の投与中止に至った有害事象を含む安全性の要約(主要評価項目、APaT)
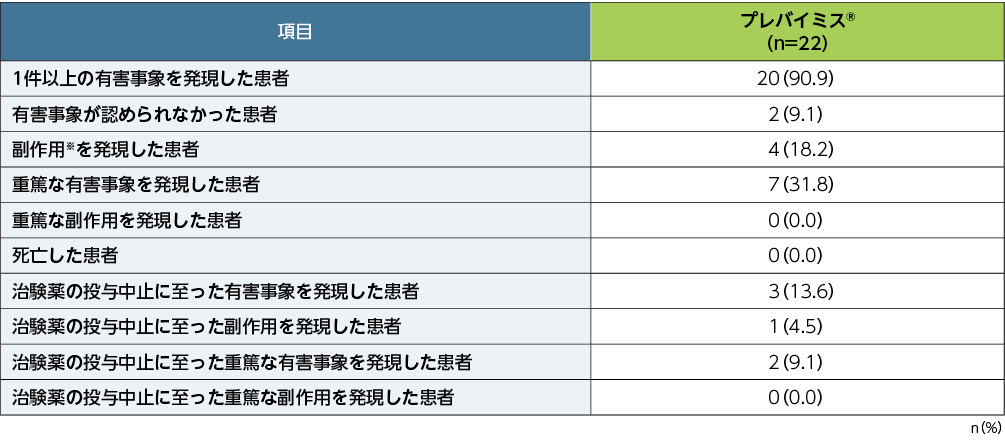
※: 治験担当医師が治験薬との因果関係ありと判定した有害事象を副作用と定義した。有害事象の評価期間は、割付け以降治験薬最終投与後14日まで収集した(治験薬投与期)。また、重篤な副作用については、治験薬投与期として規定した期間外であっても収集することとした。
2. 移植後52週以内の中央判定によるCMV感染症※1を発症した患者の割合(副次評価項目)
移植後52週以内に中央判定によるCMV感染症を発症した患者は、21例中2例(9.5%)でした(OFアプローチ。52週来院時のデータ欠測例と52週以内の試験中止例は非無効例)。
■移植後52週以内の中央判定によるCMV感染症※1を発症した患者の割合(副次評価項目、FAS)
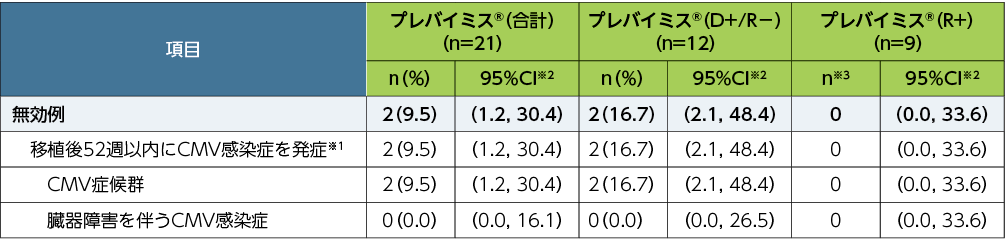
欠損値の処理法:Observed Failure approach(OFアプローチ)。OFアプローチでは、何らかの理由で早期に試験を中止した患者を非無効例として扱った。
※1:CMV感染症(臓器障害を伴うCMV感染症とCMV症候群の2種類を含むものとした)は、中央判定として独立した臨床判定委員会が確認した。CMV症候群は、血中でCMVが確認され(ウイルス分離/迅速培養/抗原血症法/核酸検査)、以下の基準のうち2項目以上に該当するものとした;①2日以上継続する38℃以上の発熱、②倦怠感あるいは疲労の新規発症又は増悪、③24時間以上の間隔で2回測定した結果に基づく白血球減少症又は好中球減少症、④異型リンパ球が5%以上、⑤血小板減少症、⑥ALT又はASTが基準値上限の2倍以上
※2:Clopper and Pearson法による正確二項検定を用いて算出した。
※3:症例数が10例未満の場合には例数のみの記載とした。
プレバイミス®(合計)にはすべての血清の状態(D+/R−、D+/R+及びD−/R+)が含まれる。
3. 移植後52週以内の中央判定によるCMV感染症※1を発症又は抗CMV療法を開始した患者の割合(副次評価項目)
移植後52週以内に中央判定によるCMV感染症を発症又は抗CMV療法を開始した患者は、21例中4例(19.0%)でした(OFアプローチ。52週来院時のデータ欠測例と52週以内の試験中止例は非無効例)。
■移植後52週以内の中央判定によるCMV感染症※1を発症又は抗CMV療法を開始した患者の割合(副次評価項目、FAS)
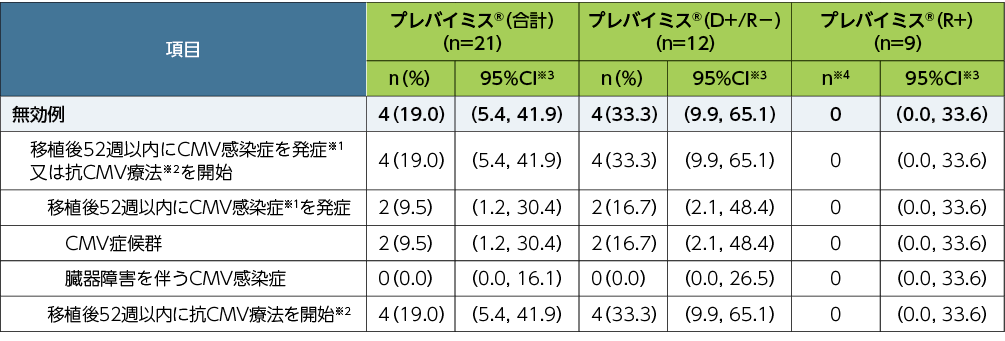
欠損値の処理法:Observed Failure approach(OFアプローチ)。OFアプローチでは、何らかの理由で早期に試験を中止した患者を非無効例として扱った。
※1:CMV感染症(臓器障害を伴うCMV感染症とCMV症候群の2種類を含むものとした)は、中央判定として独立した臨床判定委員会が確認した。CMV症候群は、血中でCMVが確認され(ウイルス分離/迅速培養/抗原血症法/核酸検査)、以下の基準のうち2項目以上に該当するものとした;①2日以上継続する38℃以上の発熱、②倦怠感あるいは疲労の新規発症又は増悪、③24時間以上の間隔で2回測定した結果に基づく白血球減少症又は好中球減少症、④異型リンパ球が5%以上、⑤血小板減少症、⑥ALT又はASTが基準値上限の2倍以上
※2:抗CMV療法の開始は以下のいずれかに基づく:①CMV抗原血症法陽性(治験実施医療機関で測定)、②定量可能なCMV DNA血症(治験実施医療機関で測定)
※3:Clopper and Pearson法による正確二項検定を用いて算出した。
※4:症例数が10例未満の場合には例数のみの記載とした。
プレバイミス®(合計)にはすべての血清の状態(D+/R−、D+/R+及びD−/R+)が含まれる。
4. 移植後28週及び52週以内の定量可能なCMV DNA血症※1がみられた患者の割合(副次評価項目)
移植後28週以内に定量可能なCMV DNA血症は21例中1例(4.8%)、移植後52週以内に定量可能なCMV DNA血症は21例中5例(23.8%)にみられました。いずれの症例もD+/R−でした。
■移植後28週及び52週以内の定量可能なCMV DNA血症※1がみられた患者の割合(副次評価項目、FAS)
移植後28週以内の定量可能なCMV DNA血症※1(副次評価項目、FAS)

※1:定量可能なCMV DNA血症は、中央検査機関で測定した結果に基づいて、測定値(≧137IU/mL)を伴うCMVの検出と定義した。
※2:Clopper and Pearson法による正確二項検定を用いて算出した。
※3:症例数が10例未満の場合には例数のみの記載とした。
プレバイミス®(合計)にはすべての血清の状態(D+/R−、D+/R+及びD−/R+)が含まれる。
移植後52週以内の定量可能なCMV DNA血症※1(副次評価項目、FAS)
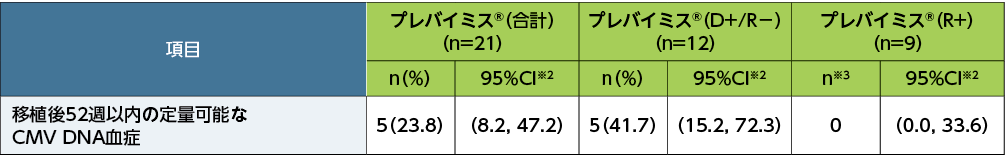
※1:定量可能なCMV DNA血症は、中央検査機関で測定した結果に基づいて、測定値(≧137IU/mL)を伴うCMVの検出と定義した。
※2:Clopper and Pearson法による正確二項検定を用いて算出した。
※3:症例数が10例未満の場合には例数のみの記載とした。
プレバイミス®(合計)にはすべての血清の状態(D+/R−、D+/R+及びD−/R+)が含まれる。
5. 安全性
副作用※発現率
移植後28週までの副作用は、22例中4例(18.2%)に認められました。頻度の高かった副作用(発現頻度1%以上)は、白血球減少症、下痢、悪心、血中アルカリホスファターゼ増加各1例(4.5%)でした。重篤な副作用は認められませんでした。投与中止に至った副作用は、白血球減少症1例(4.5%)でした。
本試験において、死亡に至った副作用は認められませんでした。
■第Ⅲ相国内試験(042試験)における副作用発現率(APaT)
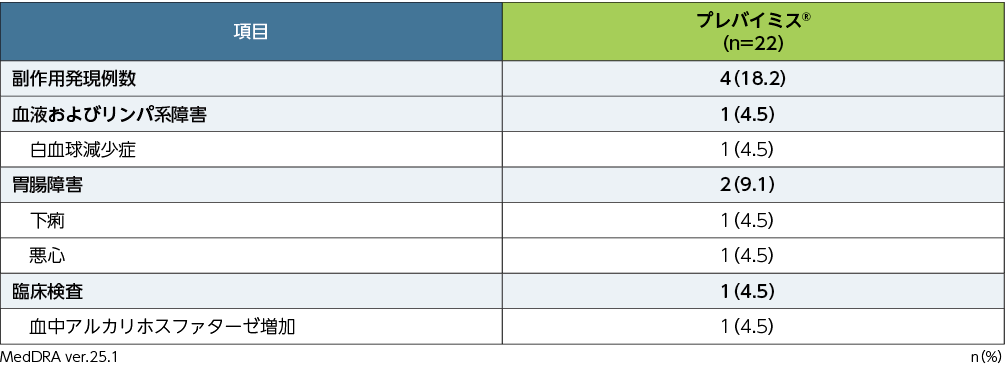
すべての患者は該当する群・分類ごとに1回ずつ集計した。
※:治験担当医師が治験薬との因果関係ありと判定した有害事象を副作用と定義した。有害事象の評価期間は、割付け以降治験薬最終投与後14日まで収集した(治験薬投与期)。また、重篤な副作用については、治験薬投与期として規定した期間外であっても収集することとした。
関連コンテンツ


