成人<臓器移植> 第Ⅲ相海外共同試験(002試験)
臨床成績
成人<臓器移植>
第Ⅲ相海外共同試験(002試験)1、2)
非日本人成人腎移植患者を対象にCMV感染症の予防*を目的としてプレバイミス®を投与した際の有効性及び安全性をバルガンシクロビルと比較する二重盲検無作為化実薬対照第Ⅲ相試験(検証試験)
1)適応追加(臓器移植)承認時評価資料(第Ⅲ相海外共同試験:002試験)
2)Limaye AP et al. JAMA. 2023; 330(1): 33-42. 利益相反:本試験はMSDより資金提供を受けている。
*:プレバイミス®の効能又は効果は、「下記におけるサイトメガロウイルス感染症の発症抑制 〇同種造血幹細胞移植 〇臓器移植」である。
試験概要
主要目的:
移植後52週以内の中央判定によるCMV感染症を発症した患者の割合に基づき、プレバイミス®の有効性をバルガンシクロビルと比較する。
対象:
CMV抗体陽性ドナー(D+)から腎移植を受けたCMV抗体陰性レシピエント(R−)の成人(D+/R−)で、移植後0日(移植当日)から7日以内の成人患者601例(無作為化症例)
試験デザイン:
海外多施設共同二重盲検無作為化実薬対照試験(日本人を含まない)[検証試験]
投与方法:
対象を導入時に強力な細胞溶解作用を有する抗リンパ球免疫療法の実施の有無で層別して1:1の比でプレバイミス®群又はバルガンシクロビル群のいずれかに無作為に割り付け、移植後7日以内にプレバイミス®経口投与480mg、もしくは静脈内投与480mg又は240mg(シクロスポリン併用時には経口・静脈内投与いずれも240mg)又はバルガンシクロビル900mg(静脈内投与の場合にはガンシクロビル**5mg/kg)を、1日1回経口又は静脈内投与にて開始し移植後28週まで継続した。プレバイミス®群の患者には単純ヘルペスウイルス及び水痘帯状疱疹ウイルスの予防のためアシクロビルを投与し、バルガンシクロビル群の患者にはアシクロビルのプラセボを投与した。バルガンシクロビル、ガンシクロビル及びアシクロビルの用量は患者の腎機能に合わせて調整した。投与完了後、移植後52週まで追跡して遅発性のCMV感染症を評価した。

※1:アシクロビルは、経口投与時に400mgを12時間ごと、静脈内投与時には250mg/m2を1日2回、併用する。
**:国内未承認の効能又は効果
**:ガンシクロビルの効能又は効果は、「下記におけるサイトメガロウイルス感染症 〇後天性免疫不全症候群 〇臓器移植(造血幹細胞移植も含む) 〇悪性腫瘍」である。
評価項目:
【主要評価項目】 移植後52週以内の中央判定によるCMV感染症※2を発症した患者の割合(検証的解析項目)
【副次評価項目】 移植後28週以内の中央判定によるCMV感染症※2を発症した患者の割合
移植後52週以内の中央判定によるCMV感染症※2がみられるまでの期間
【探索的評価項目】 移植後28週及び52週以内の定量可能なCMV DNA血症(中央検査機関で測定)がみられた患者の割合
移植後52週までの予防不成功例におけるプレバイミス®に対する耐性ウイルス 等
※2:CMV感染症は、「臓器障害を伴うCMV感染症」及び「CMV症候群」の2種類を含むものとした。
CMV症候群は、血中でCMVが確認され(ウイルス分離/迅速培養/抗原血症法/核酸検査)、以下の基準のうち2項目以上に該当するものとした;①2日以上継続する38℃以上の発熱、②倦怠感あるいは疲労の新規発症又は増悪、③24時間以上の間隔で2回測定した結果に基づく白血球減少症又は好中球減少症、④異型リンパ球が5%以上、⑤血小板減少症、⑥ALT又はASTが基準値上限の2倍以上
【安全性】 有害事象、臨床検査(血液学的検査、血液生化学的検査)、バイタルサイン、心電図 等
安全性評価項目として、白血球減少及び好中球減少を評価した。
解析計画:
- 有効性の主要解析対象集団は、無作為割付け後に治験薬の投与を1回以上受け、D+/R−であり、投与開始1日目に中央検査機関の測定でCMV DNAが検出されないすべての患者から構成される最大の解析対象集団[FAS(Full analysis set)]とした。
- 安全性の解析対象集団は、無作為割付け後に治験薬の投与を1回以上受けたすべての患者から構成されるAPaT(All Participants as Treated)とした。
- 有効性の主要解析は、主要評価項目(移植後52週以内の中央判定によるCMV感染症を発症した患者の割合)におけるバルガンシクロビルに対するプレバイミス®の非劣性を検証することであり、導入時に強力な細胞溶解作用を有する抗リンパ球免疫療法の実施の有無を層として調整したMantel-Haenszel法を用いて、2群間の差とその両側95%信頼区間(CI)を算出し、95%CIの上限が10%以下である場合に非劣性が検証されることとし、非劣性が検証された場合、95%CIの上限が0未満であれば、プレバイミス®のバルガンシクロビルに対する優越性が検証されることとした。
- 副次評価項目である移植後28週以内に中央判定によるCMV感染症を発症した患者の割合についても、主要評価項目と同様の方法を用いて2群間の差とその95%CIを算出した。
- 移植後52週以内に中央判定によるCMV感染症がみられるまでの期間について、ノンパラメトリックな方法であるKaplan-Meier法で推定し、投与群別にKaplan-Meier曲線をプロットし、導入時に強力な細胞溶解作用を有する抗リンパ球免疫療法の実施の有無で調整した層別ログランク検定を用いて群間差に関する両側p値(名目上のp値)を算出した。
- 欠測値(非完了例)は非無効例として扱った[OF(Observed Failure)アプローチ]。欠測値(非完了例)を無効例とする感度分析も実施した[NC=F(Non-Completer=Failure)アプローチ]。
- 試験中止/試験完了例はその時点で打切りとした。
解析対象例数:
有効性解析対象例数(FAS) :586例(プレバイミス®群289例、バルガンシクロビル群297例)
安全性解析対象例数(APaT): 589例(プレバイミス®群292例、バルガンシクロビル群297例)
(うち注射剤の投与を1回以上受けた患者:プレバイミス®群3例、バルガンシクロビル群2例)
患者背景
■患者背景(APaT)
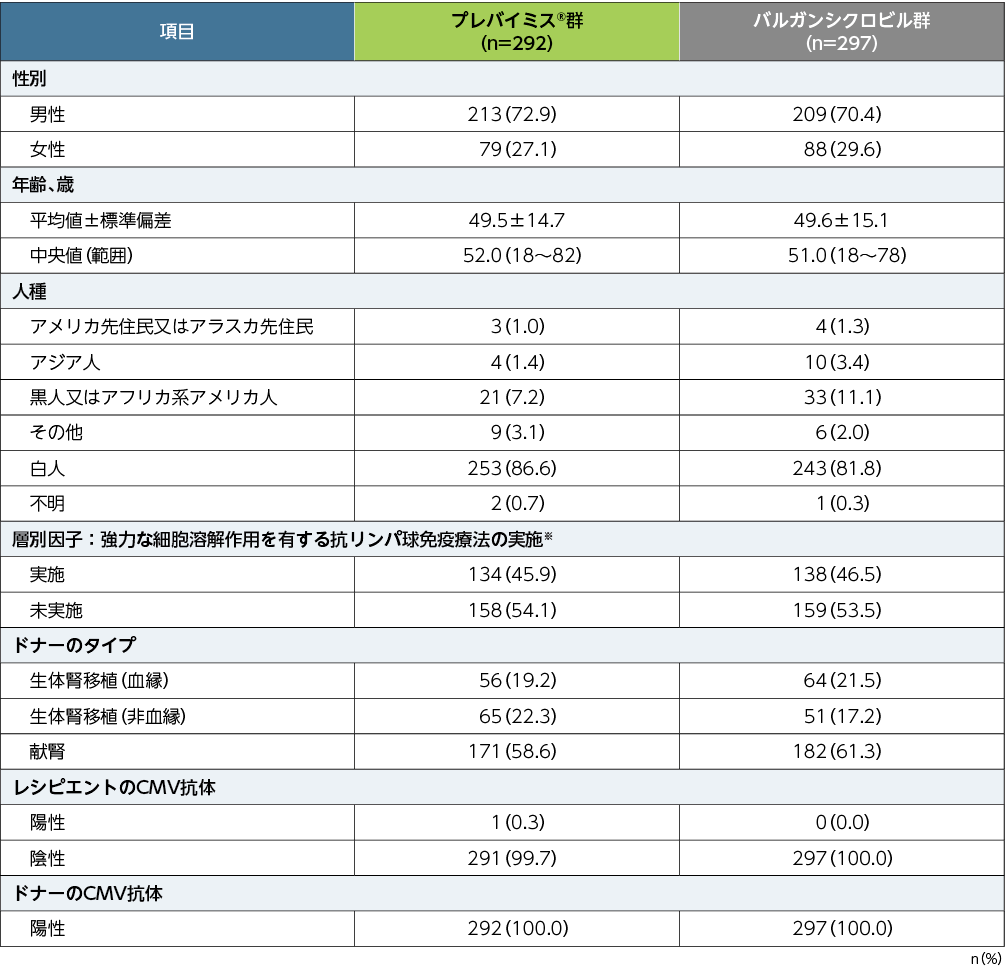
※:強力な細胞溶解作用を有する抗リンパ球免疫療法とは、抗胸腺細胞グロブリン、アレムツズマブ、又はmuromonab-CD3のいずれかによる治療の実施である。(いずれも本邦承認外の効能又は効果)
1. 移植後52週以内の中央判定によるCMV感染症※1を発症した患者の割合(主要評価項目、検証的解析結果)※2
移植後52週以内に中央判定によるCMV感染症を発症した患者の割合は、プレバイミス®群10.4%(30/289例)及びバルガンシクロビル群11.8%(35/297例)で、2群間の差は−1.4%(95%CI:−6.5%, 3.8%)であり、プレバイミス®群のバルガンシクロビル群に対する非劣性が示されましたが、95%CIの上限が0未満ではなかったため優越性は検証されませんでした[層(導入時に強力な細胞溶解作用を有する抗リンパ球免疫療法の実施/未実施)で調整したMantel-Haenszel法(各層の2群の症例数の調和平均で重み付け)により算出した2群間の差(プレバイミス®群−バルガンシクロビル群)の両側95%CIの上限が10%以下である場合に非劣性が検証されることとし、非劣性が検証された場合、95%CIの上限が0未満であれば、プレバイミス®のバルガンシクロビルに対する優越性が検証されることとした]。
■移植後52週以内の中央判定によるCMV感染症※1を発症した患者の割合(非劣性の検証、検証的解析結果、主要評価項目※2、FAS)
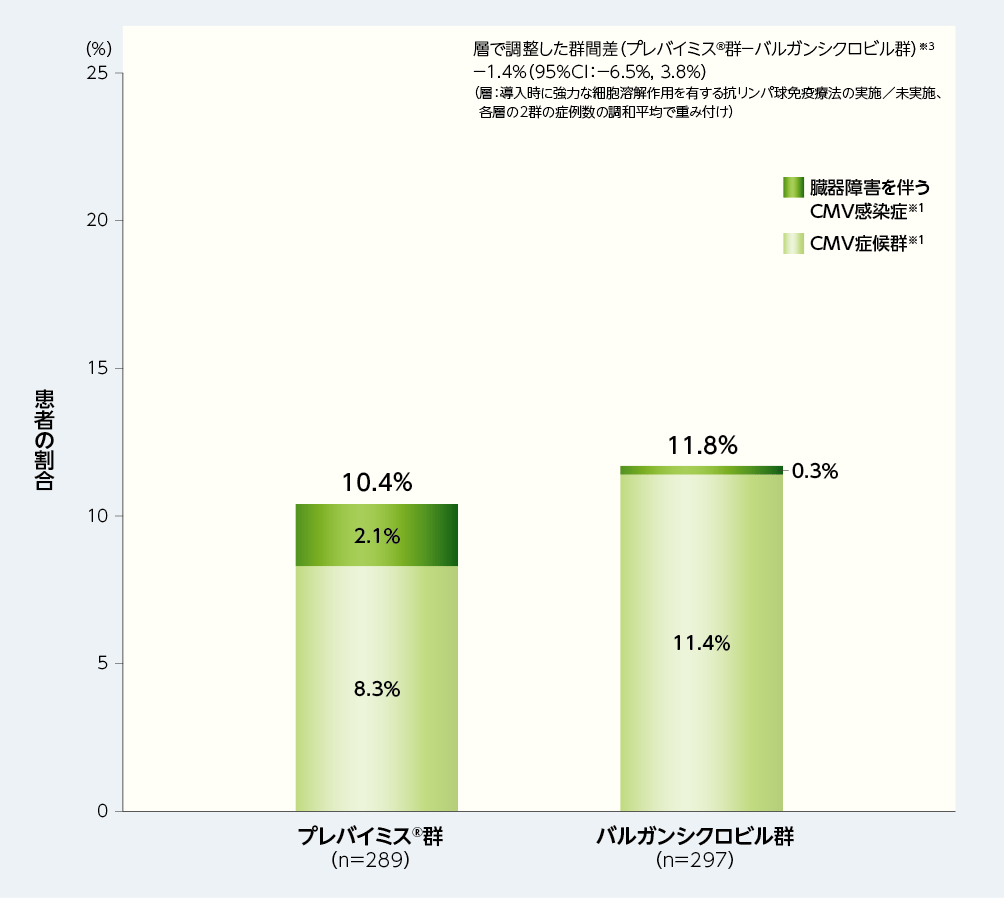
※1:CMV感染症(臓器障害を伴うCMV感染症とCMV症候群の2種類を含むものとした)は、中央判定として独立した盲検下の臨床判定委員会が確認した。CMV症候群は、血中でCMVが確認され(ウイルス分離/迅速培養/抗原血症法/核酸検査)、以下の基準のうち2項目以上に該当するものとした;①2日以上継続する38℃以上の発熱、②倦怠感あるいは疲労の新規発症又は増悪、③24時間以上の間隔で2回測定した結果に基づく白血球減少症又は好中球減少症、④異型リンパ球が5%以上、⑤血小板減少症、⑥ALT又はASTが基準値上限の2倍以上
※2:主要評価項目の解析においては、Observed Failure approach(OFアプローチ)を用いて、何らかの理由により試験を早期に中止した患者(非完了例)は非無効例とみなした。
※3:層(導入時に強力な細胞溶解作用を有する抗リンパ球免疫療法の実施/未実施)で調整したMantel-Haenszel法(各層の2群の症例数の調和平均で重み付け)を用いてCMV感染症が発症した患者の割合の差(プレバイミス®群−バルガンシクロビル群)の両側95%CIを算出し、95%CIの上限が10%以下である場合にバルガンシクロビル群に対するプレバイミス®群の非劣性が検証されることとし、非劣性が検証された場合、95%CIの上限が0未満であれば、プレバイミス®のバルガンシクロビルに対する優越性が検証されることとした。
注:プレバイミス®群はアシクロビルと併用投与した。バルガンシクロビル群はアシクロビルのプラセボと併用投与した。
2. 移植後28週以内の中央判定によるCMV感染症※1を発症した患者の割合(副次評価項目)※2
移植後28週以内に中央判定によるCMV感染症を発症した患者の割合(内訳はすべてCMV症候群であった)は、プレバイミス®群0.0%(0/289例)及びバルガンシクロビル群1.7%(5/297例)でした[群間差:−1.7%(95%CI:−3.4%, 0.1%)]。
■移植後28週以内の中央判定によるCMV感染症※1を発症した患者の割合(副次評価項目※2、FAS)
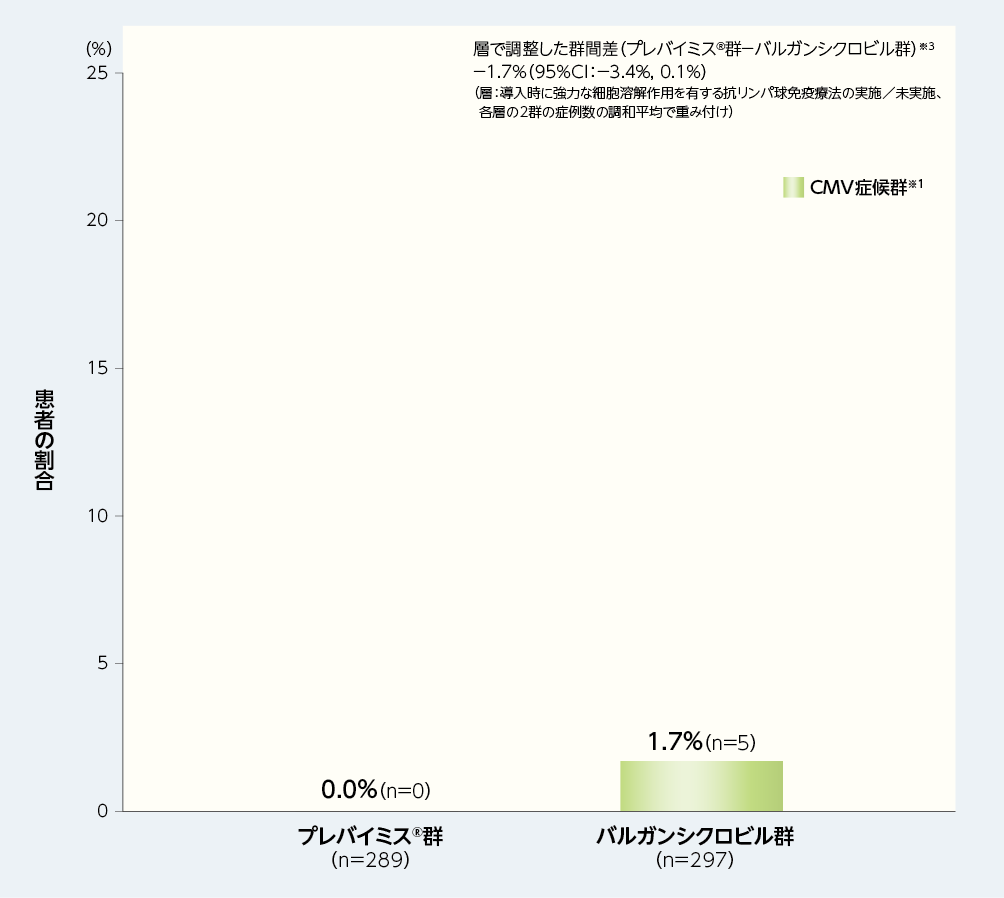
※1:CMV感染症(臓器障害を伴うCMV感染症とCMV症候群の2種類を含むものとした)は、中央判定として独立した盲検下の臨床判定委員会が確認した。CMV症候群は、血中でCMVが確認され(ウイルス分離/迅速培養/抗原血症法/核酸検査)、以下の基準のうち2項目以上に該当するものとした;①2日以上継続する38℃以上の発熱、②倦怠感あるいは疲労の新規発症又は増悪、③24時間以上の間隔で2回測定した結果に基づく白血球減少症又は好中球減少症、④異型リンパ球が5%以上、⑤血小板減少症、⑥ALT又はASTが基準値上限の2倍以上
※2:主要評価項目と同様に、Observed Failure approach(OFアプローチ)を用いて、何らかの理由により試験を早期に中止した患者(非完了例)は非無効例とみなした。
※3:層(導入時に強力な細胞溶解作用を有する抗リンパ球免疫療法の実施/未実施)で調整したMantel-Haenszel法(各層の2群の症例数の調和平均で重み付け)を用いてCMV感染症が発症した患者の割合の差(プレバイミス®群−バルガンシクロビル群)の両側95%CIを算出した。
注:プレバイミス®群はアシクロビルと併用投与した。バルガンシクロビル群はアシクロビルのプラセボと併用投与した。
3. 移植後52週以内の中央判定によるCMV感染症※1がみられるまでの期間(副次評価項目)
移植後52週以内にCMV感染症がみられるまでの期間を、Kaplan-Meier法により評価した結果、移植後52週時点のCMV感染症の非発症率は、プレバイミス®群88.2%及びバルガンシクロビル群87.1%であり、バルガンシクロビル群に対するプレバイミス®群のハザード比は0.90(95%CI:0.56, 1.47)でした。
■移植後52週以内の中央判定によるCMV感染症※1がみられるまでの期間(副次評価項目、FAS)
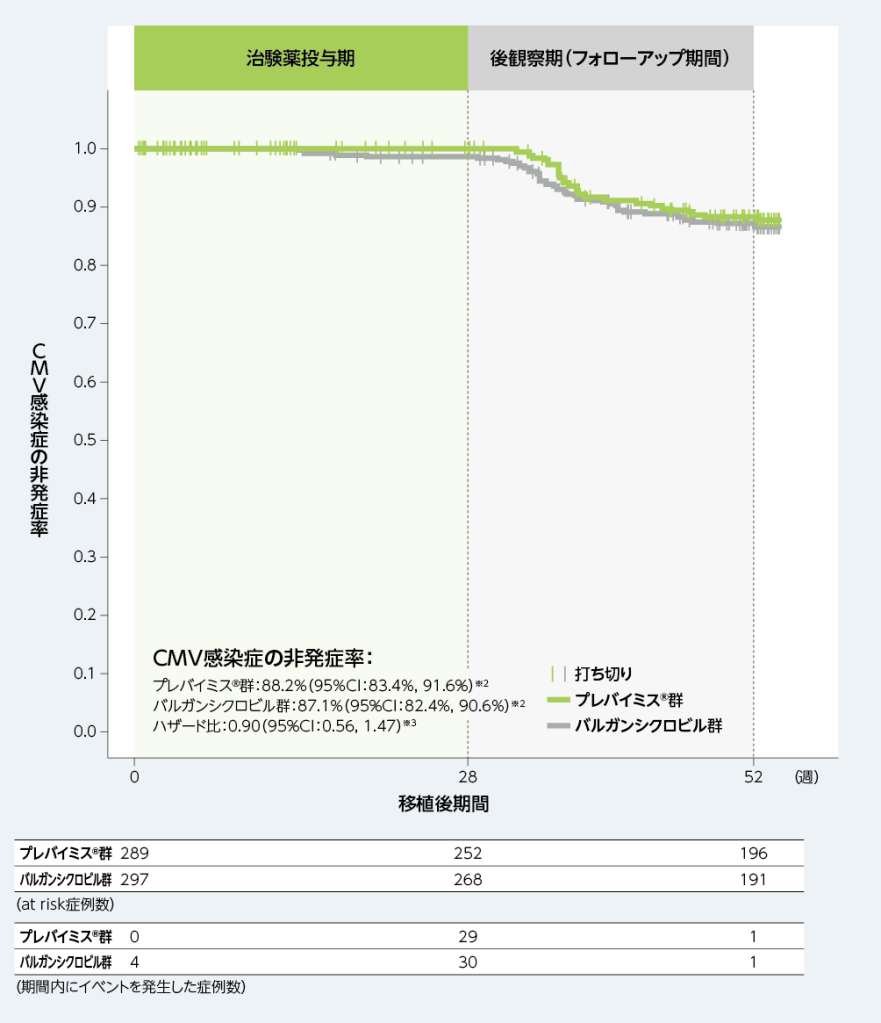
※1: CMV感染症(臓器障害を伴うCMV感染症とCMV症候群の2種類を含むものとした)は、中央判定として独立した盲検下の臨床判定委員会が確認した。CMV症候群は、血中でCMVが確認され(ウイルス分離/迅速培養/抗原血症法/核酸検査)、以下の基準のうち2項目以上に該当するものとした;①2日以上継続する38℃以上の発熱、②倦怠感あるいは疲労の新規発症又は増悪、③24時間以上の間隔で2回測定した結果に基づく白血球減少症又は好中球減少症、④異型リンパ球が5%以上、⑤血小板減少症、⑥ALT又はASTが基準値上限の2倍以上
※2:打ち切りデータを考慮したKaplan-Meier法を用いて推定した。データは最終評価時に打ち切られた。
※3: 共変量としての強力な細胞溶解作用を有する抗リンパ球免疫療法の実施/未実施で層別化した投与群を用いたEfron’s method of tie handlingによるCox回帰モデルに基づいて算出した。ハザード比<1は、プレバイミス®群で良好なことを示す。
注:プレバイミス®群はアシクロビルと併用投与した。バルガンシクロビル群はアシクロビルのプラセボと併用投与した。
4. 移植後28週及び52週以内の定量可能なCMV DNA血症※がみられた患者の割合(探索的評価項目)
移植後28週以内に定量可能なCMV DNA血症がみられた患者の割合は、プレバイミス®群2.1%(6/289例)及びバルガンシクロビル群8.8%(26/297例)でした。
移植後52週以内に定量可能なCMV DNA血症がみられた患者の割合は、プレバイミス®群31.8%(92/289例)及びバルガンシクロビル群37.7%(112/297例)でした。
■移植後28週及び52週以内の定量可能なCMV DNA血症※がみられた患者の割合(探索的評価項目、FAS)
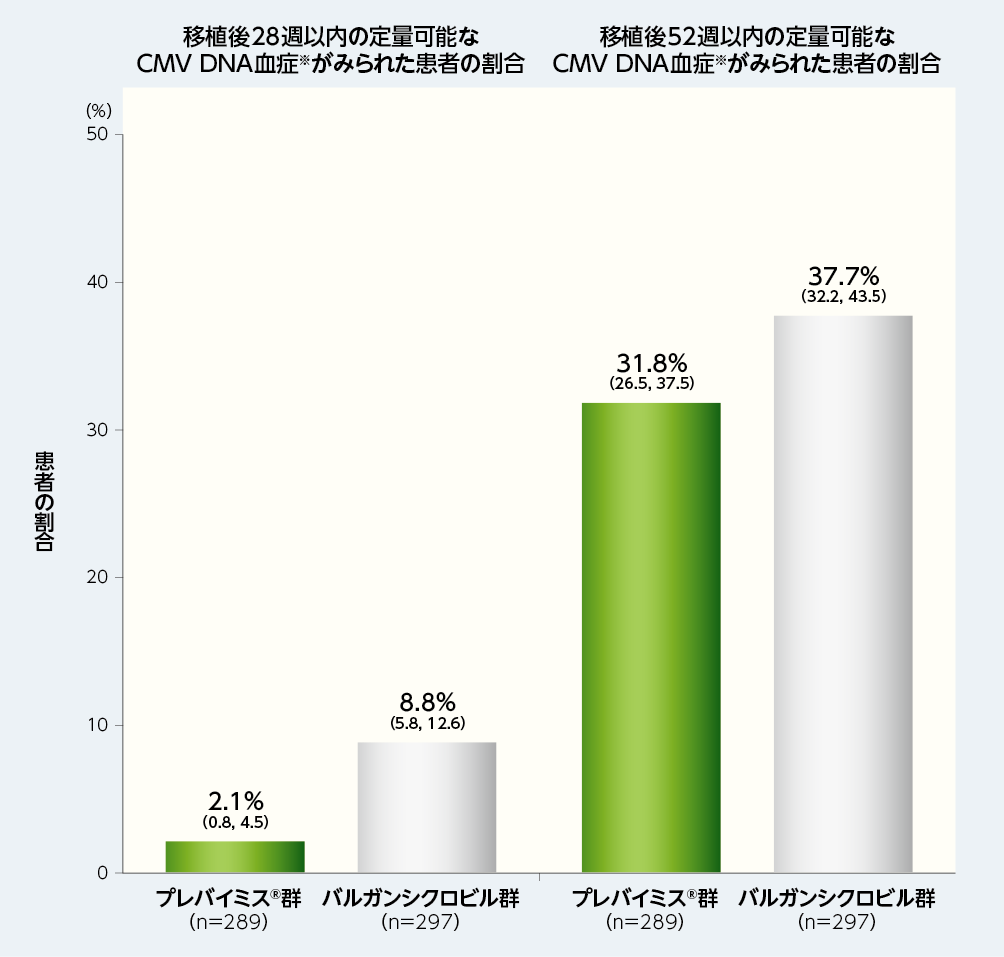
図内の数値は、患者の割合(95%CI)を示した。
※:CMV DNA血症は、中央検査機関で測定した。中央検査室の定量下限は137IU/mLであった。
注:プレバイミス®群はアシクロビルと併用投与した。バルガンシクロビル群はアシクロビルのプラセボと併用投与した。
5. 移植後52週までの予防不成功例におけるプレバイミス®に対する耐性ウイルス(探索的評価項目)
本試験においてプレバイミス®投与により、検証された測定限界を超える、プレバイミス®に対する既知の耐性変異は認められませんでした。バルガンシクロビルに対する既知の耐性変異の検出頻度は、pUL54ではプレバイミス®群0%(0/52例)、バルガンシクロビル群3.0%(2/66例)、pUL97ではプレバイミス®群3.8%(2/52例)、バルガンシクロビル群10.6%(7/66例)でした。
■CMV感染症又は早期中止での来院時にみられた耐性変異※(探索的評価項目、APaT)
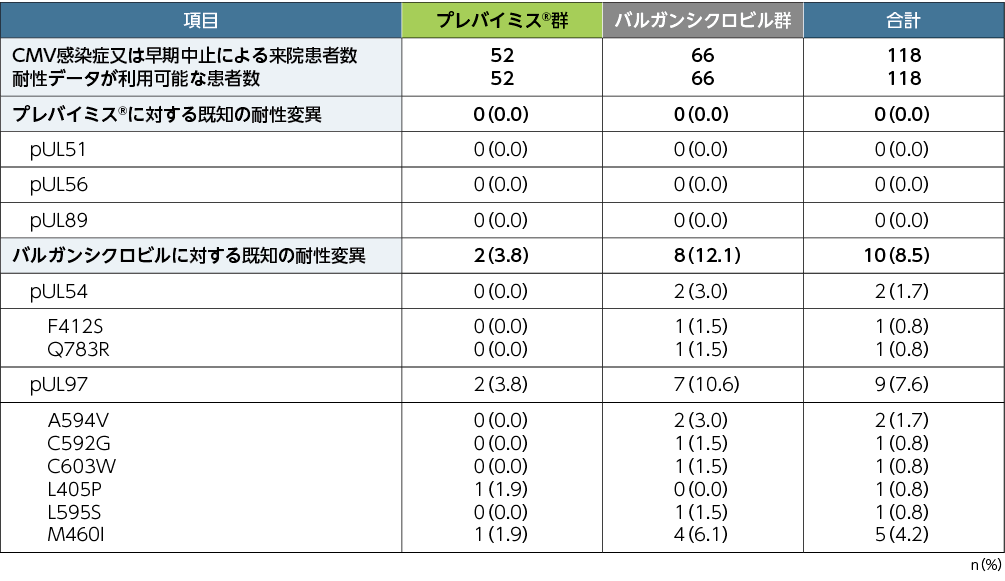
※:読み取った全シーケンスの5%以上の頻度で検出されたアミノ酸置換を有する変異を報告した。
注:プレバイミス®群はアシクロビルと併用投与した。バルガンシクロビル群はアシクロビルのプラセボと併用投与した。
6. 安全性
副作用※発現率
移植後28週までの副作用は、プレバイミス®群で292例中58例(19.9%)、バルガンシクロビル群で297例中104例(35.0%)に認められました。両群で頻度の高かった副作用(発現頻度1%以上)は、プレバイミス®群で白血球減少症20例(6.8%)、好中球減少症6例(2.1%)、振戦4例(1.4%)、薬物濃度治療量以上、白血球数減少各3例(1.0%)、バルガンシクロビル群で白血球減少症68例(22.9%)、好中球減少症24例(8.1%)、白血球数減少12例(4.0%)、貧血5例(1.7%)、発熱性好中球減少症、下痢各3例(1.0%)でした。重篤な副作用は、プレバイミス®群で白血球減少症、好中球減少症各2例、発熱性好中球減少症、汎血球減少症、心房細動、下痢各1例、バルガンシクロビル群で白血球減少症6例、発熱性好中球減少症、白血球数減少各3例、好中球減少症2例、悪心、低リン血症各1例でした。投与中止に至った副作用は、プレバイミス®群8例(2.7%)で、内訳は好中球減少症4例、白血球減少症3例、汎血球減少症2例、バルガンシクロビル群26例(8.8%)で、内訳は白血球減少症16例、好中球減少症4例、白血球数減少3例、下痢、悪心、嚥下障害各1例でした。
本試験において、死亡に至った副作用は認められませんでした。
■第Ⅲ相海外共同試験(002試験)における副作用発現率(いずれかの群で発現頻度≧1%、APaT)
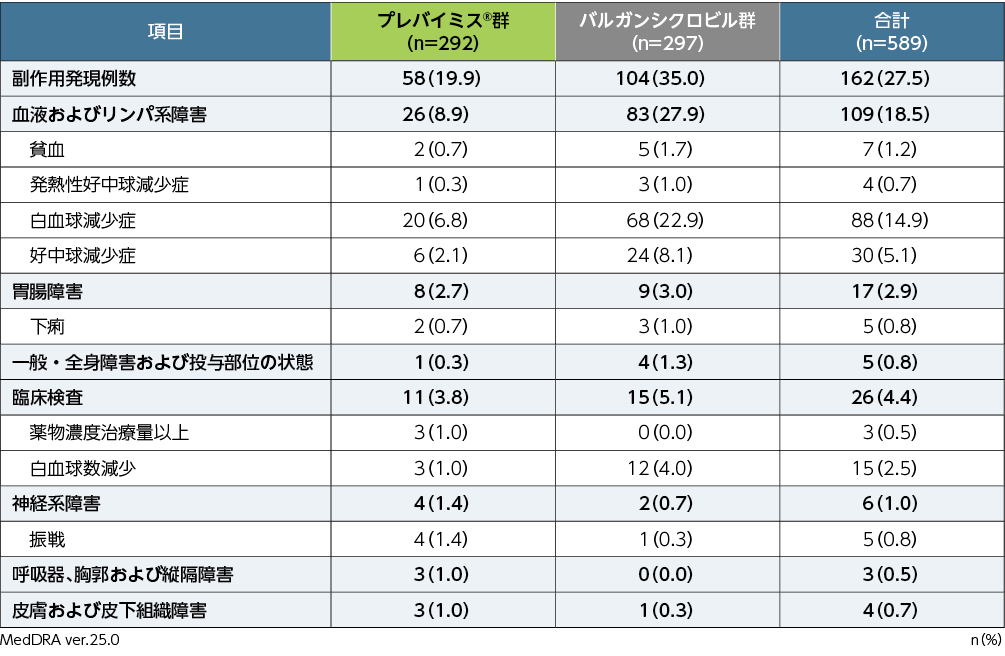
すべての患者は該当する群・分類ごとに1回ずつ集計した。
※:治験担当医師が盲検下で治験薬との因果関係ありと判定した有害事象を副作用と定義した。有害事象の評価期間は、割付け以降治験薬最終投与後14日まで収集した(治験薬投与期)。また、重篤な副作用については、治験薬投与期として規定した期間外であっても収集することとした。
関連コンテンツ


