〈レンビマ®併用〉国際共同第Ⅲ相試験〈CLEAR試験〉(KEYNOTE-581/307試験)(日本人集団)
国際共同第Ⅲ相試験〈CLEAR試験(KEYNOTE-581/307試験)〉(日本人集団)
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-581/E7080-307試験)
Motzer R et al. N Engl J Med 2021; 384: 1289-1300#1
#1 CLEAR試験(KEYNOTE-581/E7080-307試験)はエーザイ株式会社・MSD社の資金提供により行われた。Robert Motzerはエーザイ株式会社・MSD社から顧問料などを受領している。その他の著者に同社から顧問料などを受領している者が含まれる。本試験はアカデミックアドバイザーとエーザイ株式会社・MSD社の社員によりデザインされた。著者のうち、Rodolfo F. PeriniはMSD社、Alan D. Smith、Corina E. Dutcus、Lea Dutta、Kalgi Mody、Dongyuan Xingはエーザイ株式会社の社員である。
※日本人集団については、事前規定されていないが、評価資料として承認時に評価されたため記載した
本併用療法は、一部承認外の効能又は効果、用法及び用量による臨床試験の成績も含めた臨床データパッケージで評価され、承認されました。本試験におけるレンビマ®+エベロリムス併用群のデータは国内未承認のため削除しています。
試験概要
【目的】
化学療法未治療の根治切除不能又は転移性腎細胞癌患者におけるキイトルーダ®+レンビマ®併用又はレンビマ®+エベロリムス併用とスニチニブ単独の有効性及び安全性を比較検討する。
【デザイン】
国際共同無作為化非盲検第Ⅲ相試験[優越性検証試験]
[第1回中間解析結果(データカットオフ日:2018年12月6日)、第2回中間解析結果(データカットオフ日:2019年11月15日)、第3回中間解析結果(データカットオフ日:2020年8月28日)、第4回中間解析結果(データカットオフ日:2021年3月31日)、最終解析結果(データカットオフ日:2022年7月31日)]
【対象】
化学療法未治療の根治切除不能又は転移性腎細胞癌患者1,069例
【方法】
キイトルーダ®+レンビマ®※併用群[キイトルーダ®200mgを3週間間隔(Q3W)で点滴静注とレンビマ®※20mgを1日1回(QD)で経口投与]、レンビマ®+エベロリムス併用群注)[レンビマ®18mgQDで経口投与とエベロリムス5mgQDで経口投与]又はスニチニブ群[50mgQDで4週間経口投与後、2週間休薬]に1:1:1の割合で無作為に割り付けた。無作為化後、盲検下独立中央判定を8週間毎に実施し、抗腫瘍効果を評価した。盲検下独立中央判定による進行(PD)の確定、許容できない毒性の発現、患者の要望、同意撤回、キイトルーダ®35回投与の完了又は治験依頼者による治験中止まで治験薬の投与を継続した。
※レンビマ®の効能又は効果、用法及び用量は以下のとおりです(一部抜粋)。
4. 効能又は効果(抜粋)
根治切除不能又は転移性の腎細胞癌
6. 用法及び用量(抜粋)
<根治切除不能又は転移性の腎細胞癌>
ペムブロリズマブ(遺伝子組換え)との併用において、通常、成人にはレンバチニブとして1日1回20mgを経口投与する。なお、患者の状態により適宜減量する。
7. 用法及び用量に関連する注意(抜粋)
<根治切除不能又は転移性の腎細胞癌>
7.6 副作用があらわれた場合は、症状、重症度等に応じて以下の基準を考慮して、本剤を減量、休薬又は中止すること。減量して投与を継続する場合には、1日1回14mg、10mg、8mg又は4mgに減量すること。
減量、休薬及び中止基準
【評価項目】
主要評価項目:無増悪生存期間(progression free survival;PFS)#
副次評価項目:全生存期間(overall survival;OS)#及び奏効率(overall response rate;ORR)#、次治療におけるPDまでの無増悪生存期間(progression free survival during next-line therapy; PFS2)、健康関連QoL(HRQoL)、安全性 など
その他の評価項目:奏効期間(duration of response;DOR)、病勢コントロール率(disease control rate;DCR)など
#検証的解析項目
【判定基準】
PFS、ORR、DORについて、RECISTガイドライン1.1版に基づき盲検下独立中央判定により評価した。PFS2は、RECISTガイドライン1.1版に基づき治験責任医師判定により評価した。HRQoLへの影響をFunctional Assessment of Cancer Therapy Kidney Symptom Index-Disease-Related Symptoms(FKSI-DRS)、European Organization for Research and Treatment of Cancer Quality of Life Questionnaire for Patients With Cancer-Core 30(EORTC QLQ-C30)及びEQ-5D-3L with the associated EuroQoL Visual Analogue Scale(EQ-VAS)を用いて評価した。
【解析計画】
解析対象集団:有効性はITT集団*5、安全性は安全性解析対象集団*6を解析対象とした。
統計手法:OS、PFS、PFS2はKaplan-Meier法を用いて推定した。OS、PFS、PFS2の群間比較は地域(西欧及び北米又はその他)及びMSKCCリスク分類(favorable、intermediate又はpoor)を層とした層別ログランク検定により評価した。ハザード比と95%信頼区間の算出は、投与群を共変量とし、地域及びMSKCCリスク分類で層別した層別Cox比例ハザードモデルにより評価した。ORRの群間差は、地域及びMSKCCリスク分類を層としたCochran-Mantel-Haenszel検定により評価した。また、年齢(<65歳、≧65歳)、性別(男性、女性)、人種(白人、アジア人)、地域(西欧及び北米、その他)、MSKCCリスク分類(favorable、intermediate、poor)、IMDCリスク分類(favorable、intermediate、poor)、ベースライン時のKPSスコア(100-90、80-70)、転移臓器数(1、2、≧3)、ベースライン時の骨転移(あり、なし)、ベースライン時の肝転移(あり、なし)、ベースライン時の肺転移(あり、なし)、PD-L1発現(CPS≧1、CPS<1)、腎摘除術歴(あり、なし)、肉腫様成分(あり、なし)についてのPFS、OS及びORRの部分集団解析を実施した。
日本人集団については、治験実施計画書に記載されていないが、OS、PFS、ORR、DORについても算出し、評価資料として承認時に評価された。
多重性の調整:本試験において、PFSは1回の中間解析及び最終解析、OSは3回の中間解析及び最終解析、ORRは1回の中間解析及び最終解析を事前に計画した。ORRの1回目の中間解析時の推定における両側0.0001の有意水準を差し引き、PFS、OS及びORRの検定の手順全体の有意水準を両側0.0499となるように厳密に制御した。これらの多重性の調整には、Maurer & Bretzのgraphical approachを用いた。他の副次及び探索的な評価項目に関しては多重性の調整は実施しない。最初に割り当てられた有意水準0.045(又は0.0049)にてPFSの帰無仮説が棄却された場合、この有意水準0.045(又は0.0049)に示される重みに従い、他の検定に再配分した。PFS、OSの中間解析及び最終解析の有意水準の配分はα消費関数にて決定した。
検定手順:
*1 MSKCC(Memorial Sloan Kettering Cancer Center)リスク分類:5つのリスク因子[Karnofsky PS80%未満、初診から治療開始までの期間(本試験の無作為化までの期間)が1年未満、ヘモグロビン値が正常範囲の下限未満、乳酸脱水素酵素が正常値上限の1.5倍超、補正血清カルシウム値が10.0mg/dL超]で構成され、MSKCCスコア0をfavorable、1-2をintermediate、3以上をpoorに分類
*2 副作用管理を目的として、段階的な減量(14mg、10mg、8mg)を可能とした
*3 4週間連日投与後、2週間休薬
*4 副作用管理を目的として、段階的な減量(37.5mg QD、25mg QD)を可能とした
*5 ITT(intention-to-treat)集団:無作為化されたすべての患者
*6 安全性解析対象集団:治験薬が1回以上投与されたすべての患者
患者背景(日本人集団)
*1 MSKCC(Memorial Sloan Kettering Cancer Center)リスク分類:5つのリスク因子[Karnofsky PS80%未満、初診から治療開始までの期間(本試験の無作為化までの期間)が1年未満、ヘモグロビン値が正常範囲の下限未満、乳酸脱水素酵素が正常値上限の1.5倍超、補正血清カルシウム値が10.0mg/dL超]で構成され、MSKCCスコア0をfavorable、1-2をintermediate、3以上をpoorに分類
*2 IMDC(International Metastatic Renal Cell Carcinoma Database Consortium)リスク分類:6つのリスク因子[Karnofsky PS80未満、初診から治療開始までの期間(本試験の無作為化までの期間)が1年未満、ヘモグロビン値が正常範囲の下限未満、補正血清カルシウム値が正常範囲の上限超、好中球数が正常範囲の上限超、血小板数が正常範囲の上限超]で構成され、IMDCスコア0をfavorable、1-2をintermediate、3-6をpoorに分類
*3 immunohistochemistry 22C3 pharmDxを用いて、CPS[combined positive score:PD-L1陽性細胞数(腫瘍細胞、リンパ球及びマクロファージ)を総腫瘍細胞数で除し、100を乗じた数値]1以上を陽性とした
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-581/E7080-307試験)
サブグループ解析:日本人集団における無増悪生存期間(PFS)
PFS中央値はキイトルーダ®+レンビマ®併用群で22.1ヵ月(95%CI:11.9, 未到達)、スニチニブ群で10.9ヵ月(95%CI:5.6, 11.2)でした。6ヵ月時点の無増悪生存率はそれぞれ82.6%(95%CI:66.9, 91.3)、65.9%(95%CI:44.5, 80.7)、12ヵ月時点の無増悪生存率はそれぞれ66.9%(95%CI:49.8, 79.3)、29.4%(95%CI:11.7, 49.9)、18ヵ月時点の無増悪生存率はそれぞれ55.5%(95%CI:38.3, 69.6)、23.6%(95%CI:7.8, 44.0)、24ヵ月時点の無増悪生存率はそれぞれ49.3%(95%CI:30.4, 65.7)、7.9%(95%CI:0.6, 28.5)でした。
■無増悪生存期間(PFS)のKaplan-Meier曲線(日本人集団)

RECISTガイドライン1.1版に基づく盲検下独立中央判定
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量としたCox比例ハザードモデルに基づく
[追跡期間(PFSの打ち切りイベントを反転させたデータを用いKaplan-Meier推定法で算出)中央値(95%CI) キイトルーダ®+レンビマ®併用群:20.3(18.4, 24.0)ヵ月、スニチニブ群:18.4(9.1, 27.7)ヵ月]
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-581/E7080-307試験)
サブグループ解析:日本人集団における全生存期間(OS)
OS中央値はキイトルーダ®+レンビマ®併用群で未到達、スニチニブ群で30.6ヵ月(95%CI:24.8, 30.6)でした。12ヵ月時点の全生存率はそれぞれ97.6%(95%CI:84.3, 99.7)、93.3%(95%CI:75.9, 98.3)、18ヵ月時点の全生存率はそれぞれ87.7%(95%CI:72.9, 94.7)、89.7%(95%CI:71.4, 96.6)、24ヵ月時点の全生存率はそれぞれ75.1%(95%CI:54.8, 87.2)、85.7%(95%CI:66.0, 94.4)でした。
■全生存期間(OS)のKaplan-Meier曲線(日本人集団)
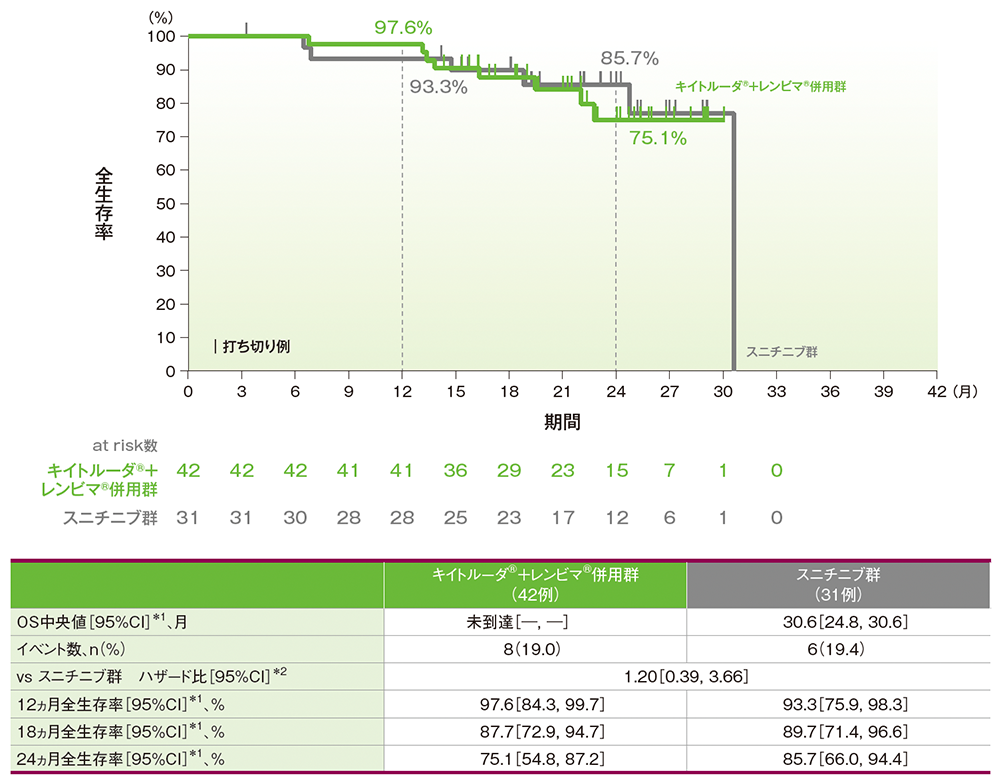
*1 打ち切りデータはproduct-limit(Kaplan-Meier)法に基づく
*2 投与群を共変量としたCox比例ハザードモデルに基づく
[追跡期間(OSの打ち切りイベントを反転させたデータを用いKaplan-Meier推定法で算出)中央値(95%CI) キイトルーダ®+レンビマ®併用群:23.0(19.6, 25.4)ヵ月、スニチニブ群:23.1(19.6, 25.4)ヵ月]
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-581/E7080-307試験)
サブグループ解析:日本人集団における奏効率(ORR)
ORRはキイトルーダ®+レンビマ®併用群69.0%(95%CI:55.1, 83.0)、スニチニブ群45.2%(95%CI:27.6, 62.7)でした(群間差:23.9、95%CI:1.5, 46.3)。
■奏効率(ORR:CR+PR)(日本人集団)
■ORR及び奏効期間(DOR)(日本人集団)
RECISTガイドライン1.1版に基づく盲検下独立中央判定
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-581/E7080-307試験)
安全性
キイトルーダ®+レンビマ®併用群
キイトルーダ®+レンビマ®併用群における副作用は42例全例(100%)に認められました。主な副作用(発現率20%以上)は手掌・足底発赤知覚不全症候群27例(64.3%)、甲状腺機能低下症26例(61.9%)、高血圧24例(57.1%)、下痢、蛋白尿及び発声障害 各21例(50.0%)、食欲減退17例(40.5%)、口内炎16例(38.1%)、倦怠感14例(33.3%)、血小板数減少10例(23.8%)でした。重篤な副作用は21例(50.0%)に認められ、2例以上にみられた重篤な副作用は副腎機能不全5例(11.9%)、食欲減退3例(7.1%)でした。副作用によるキイトルーダ®及びレンビマ®の投与中止は6例(14.3%)に認められ、その内訳は急性心不全、下痢、肝機能異常、膀胱炎、腎不全、肺障害、手掌・足底発赤知覚不全症候群 各1例(2.4%)でした。副作用によるキイトルーダ®の投与中止は10例(23.8%)に認められ、その内訳は急性心不全、下痢、免疫性膵炎、免疫性肝炎、肝機能異常、膀胱炎、ニューモシスチス・イロベチイ肺炎、腎不全、肺臓炎、肺障害、手掌・足底発赤知覚不全症候群 各1例(2.4%)でした。副作用によるレンビマ®の投与中止は12例(28.6%)に認められ、その内訳は心筋梗塞、急性心不全、下痢、肝機能異常、膀胱炎、心電図QT延長、リパーゼ増加、腎不全、肺障害、発疹、薬疹、多形紅斑、手掌・足底発赤知覚不全症候群 各1例(2.4%)でした。本試験の日本人集団において、キイトルーダ®+レンビマ®併用群の副作用による死亡は認められませんでした。
スニチニブ群
スニチニブ群の副作用は31例全例(100%)に認められました。主な副作用(発現率20%以上)は血小板数減少20例(64.5%)、手掌・足底発赤知覚不全症候群19例(61.3%)、味覚不全16例(51.6%)、高血圧15例(48.4%)、下痢13例(41.9%)、白血球数減少12例(38.7%)、甲状腺機能低下症及び倦怠感 各11例(35.5%)、口内炎及び発熱 各10例(32.3%)、好中球数減少9例(29.0%)、リパーゼ増加8例(25.8%)、食欲減退及び蛋白尿 各7例(22.6%)でした。重篤な副作用は10例(32.3%)に認められ、2例以上にみられた重篤な副作用は発熱3例(9.7%)でした。副作用によるスニチニブの投与中止は4例(12.9%)に認められ、その内訳は発熱、敗血症、血小板数減少、反回神経麻痺 各1例(3.2%)でした。本試験の日本人集団において、スニチニブ群の副作用による死亡は認められませんでした。
■主な副作用(いずれかの投与群で発現率5%以上)(日本人集団)
MedDRA/J v23.0、GradeはNCI CTCAE v4.03
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-581/E7080-307試験)
免疫関連など特に注目すべき有害事象
キイトルーダ®+レンビマ®併用群における免疫関連など特に注目すべき有害事象は31/42例(73.8%)に認められました。主な免疫関連など特に注目すべき有害事象(発現率10%以上)は甲状腺機能低下症26例(61.9%)、副腎機能不全6例(14.3%)でした。重篤な免疫関連など特に注目すべき有害事象は10例(23.8%)に認められ、2例以上にみられた重篤な免疫関連など特に注目すべき有害事象は副腎機能不全5例(11.9%)、肝炎2例(4.8%)でした。免疫関連など特に注目すべき有害事象によるキイトルーダ®の投与中止は3例(7.1%)に認められ、キイトルーダ®の投与中止に至った免疫関連など特に注目すべき有害事象は肝炎、膵炎及び肺臓炎 各1例(2.4%)でした。本試験の日本人集団において、免疫関連など特に注目すべき有害事象による死亡は認められませんでした。
スニチニブ群における免疫関連など特に注目すべき有害事象11/31例(35.5%)に認められました。主な免疫関連など特に注目すべき有害事象(発現率10%以上)は甲状腺機能低下症11例(35.5%)でした。本試験の日本人集団において、重篤な免疫関連など特に注目すべき有害事象、免疫関連など特に注目すべき有害事象によるスニチニブの投与中止、免疫関連など特に注目すべき有害事象による死亡は認められませんでした。
■免疫関連など特に注目すべき有害事象(日本人集団)
MedDRA/J v23.0、GradeはNCI CTCAE v4.03
承認時評価資料:国際共同第Ⅲ相試験(KEYNOTE-581/E7080-307試験)
関連コンテンツ
キイトルーダ®・悪性腫瘍関連領域情報


キイトルーダ®治療日誌:<頭頸部癌>キイトルーダ®術前補助療法
