開発の経緯
アクチビンシグナル伝達阻害剤であるエアウィン®の開発の経緯をご紹介します。
承認時評価資料:国内第Ⅲ相試験(020試験)


† いずれの投与期でも、用量調整ガイドラインに基づきエアウィン®の用量を増減する。
‡ 24週時の治験薬投与前までを主要投与期のデータとして扱った。
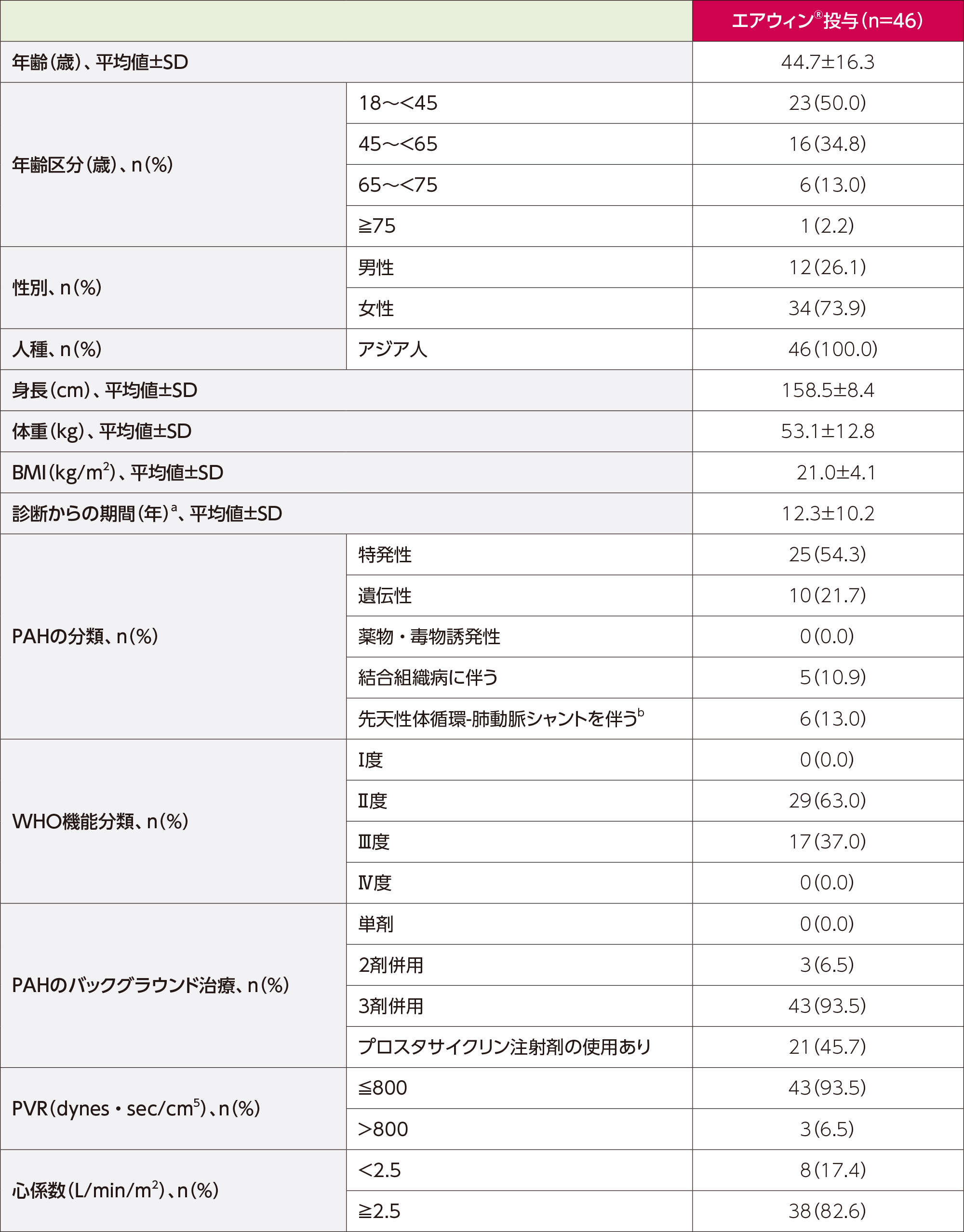
a:(治療割付け日-PAHの診断日)/365.25により算出
b:修復術施行後1年以上経過した単純性の先天性体循環-肺動脈シャントを伴うPAH
24週時のPVRのベースラインからの変化量の推定値aは−99.2dynes・sec/cm5でした。
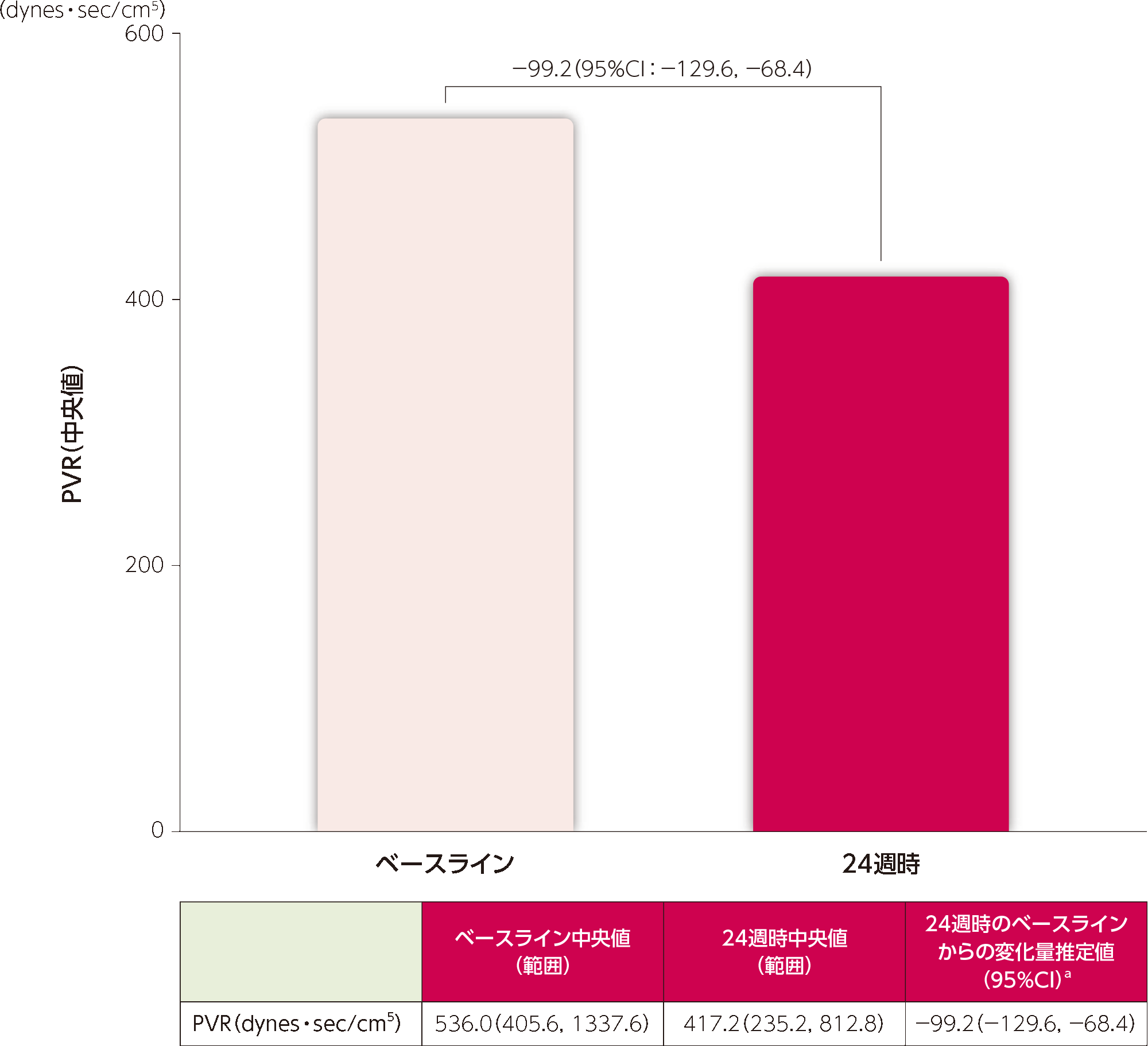
a:Hodges-Lehmann法による位置パラメータの推定値
24週時の6分間歩行距離のベースラインからの変化量の推定値aは41.8m、NT-proBNPのベースラインからの変化量の推定値aは−48.5pg/mLでした。
エアウィン®投与(n=46)
a:Hodges-Lehmann法による位置パラメータの推定値
24週時のWHO機能分類がベースラインから改善した患者割合は19.6%(9/46例)でした。

a:Clopper and Pearson法に基づく95%CI
24週時のmPAPのベースラインからの変化量の中央値は-7.5mmHgでした。

エアウィン®投与(n=46)
54週時点でデータが得られた43例†における54週時の6分間歩行距離のベースラインからの変化量及び54週時のWHO機能分類がベースラインから改善した患者割合の結果は以下の通りでした。
†54週時点での治験薬投与継続例数は42例であったが、54週時点より前に試験終了した1例で得られたEOT来院データが、54週時点の解析で使用するデータの許容範囲内に存在することになったため、sSAP(supplemental statistical analysis plan)の取扱いに従い、54週時点の評価例数としては43例となった。
● 54週時の6分間歩行距離のベースラインからの変化量の平均値(SD):52.4(51.7)m
● 54週時のWHO機能分類がベースラインから改善した患者割合:25.6%(11/43例)
副作用の発現割合は69.6%(32/46例)であり、主な副作用(発現割合5%以上)は、ヘモグロビン増加28.3%(13/46例)、鼻出血26.1%(12/46例)、頭痛17.4%(8/46例)及び毛細血管拡張症6.5%(3/46例)でした。
重篤な副作用及び治験薬の投与中止に至った副作用は認められませんでした。
データカットオフ時点までに死亡に至った副作用は認められませんでしたが、有害事象の集計対象期間(治験薬の初回投与から最終投与8週後まで)外に、治験担当医師が本剤との関連ありと判定した死亡に至った有害事象(胃腸出血)が1例報告されました。
アクチビンシグナル伝達阻害剤であるエアウィン®の開発の経緯をご紹介します。
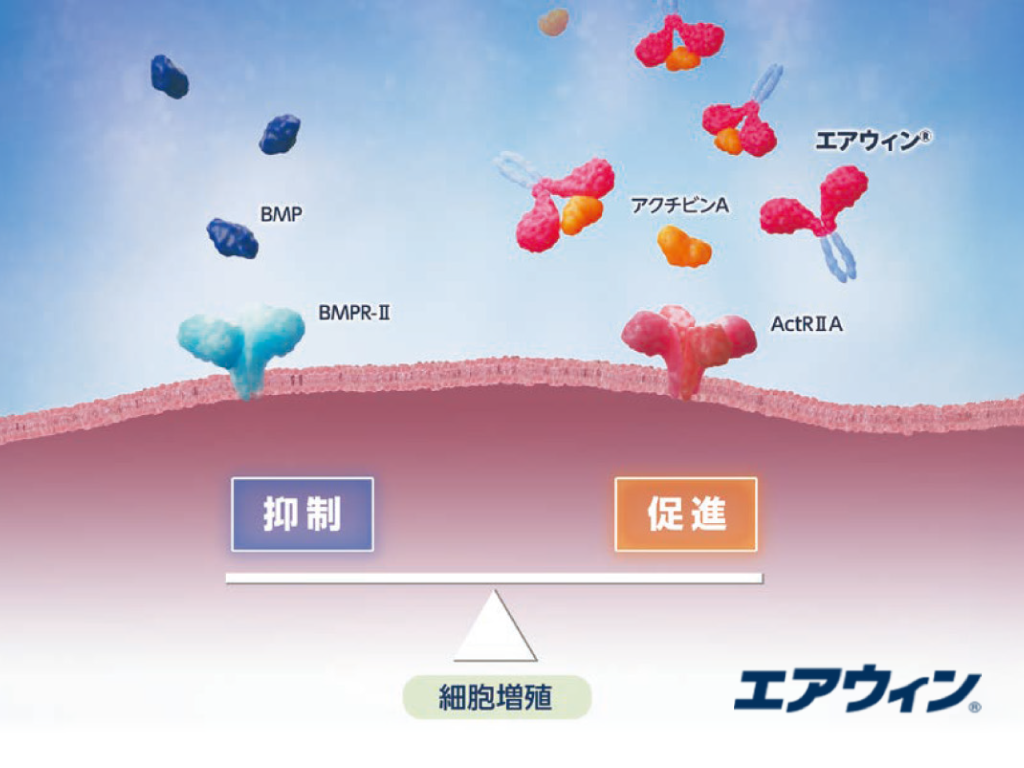
エアウィン®は肺血管拡張とは異なる経路を標的とするアクチビンシグナル伝達阻害剤です。ここでは、エアウィン®の作用機序をご紹介します。

PAHのバックグラウンド治療を受けている患者さんに、エアウィン®又はプラセボを投与した際の有効性及び安全性を評価した海外第Ⅲ相試験(STELLAR試験)をご紹介します。
WEB講演会にアクセスし「このページは閲覧を制限しています」と表示された方は こちら>>
このサイトでは、医療用医薬品を適正にご使用いただくため、医師、歯科医師及び薬剤師などの医療関係者の方を対象に、主としてMSD株式会社の医療用医薬品に関する情報を提供しています。
下記の「はい」をクリックした場合、「MSD Connect ご利用規約」及び「ウェブサイトのご利用条件」を理解したうえで、内容に同意したものとみなします。
2024年11月にご利用規約を改訂致しました。上記リンクよりご確認ください。
あなたは医療関係者ですか?