国際共同第Ⅲ相試験(052試験)
国際共同第Ⅲ相試験(052試験)
BIC/FTC/TAFによりウイルス学的抑制が得られているHIV-1感染症患者を対象とした二重盲検試験
承認時評価資料:国際共同第Ⅲ相試験(052試験)
1.試験概要
目的:BIC/FTC/TAF(ビクテグラビル/エムトリシタビン/テノホビルアラフェナミド)によりウイルス学的抑制が得られているHIV-1感染症患者を対象に、イドビンソ®配合錠に切り替えた場合の有効性及び安全性を、BIC/FTC/TAFを継続した場合と比較検討する。
試験デザイン:多施設共同、無作為化、二重盲検、実薬対照第Ⅲ相、非劣性検証試験
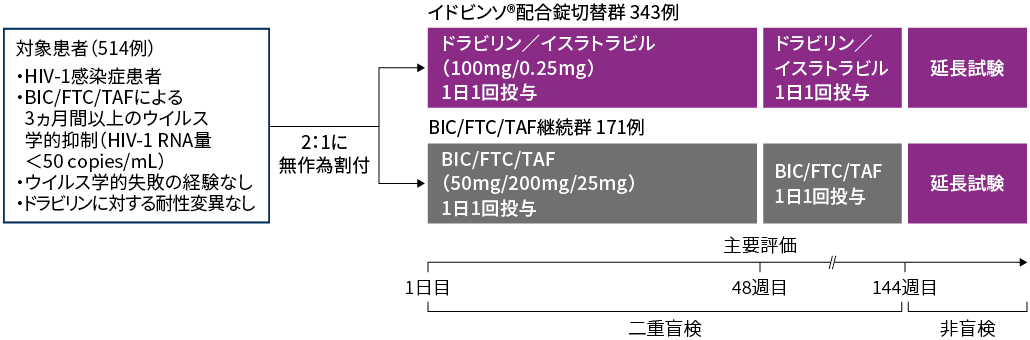
対象:BIC/FTC/TAFによりウイルス学的抑制(HIV-1 RNA量50 copies/mL未満)が3ヵ月間以上得られており、ウイルス学的失敗の経験がなく、ドラビリンに対する耐性変異が確認されていない18歳以上のHIV-1感染症患者514例※(日本人17例を含む)
※活動性HBV感染患者(HBs抗原陽性又はHBV DNA陽性)は本試験から除外
方法:対象患者を1日目にBIC 50mg/FTC 200mg/TAF 25mgの1日1回投与からイドビンソ®配合錠(ドラビリン 100mg/イスラトラビル 0.25mg)の1日1回投与に切り替えたイドビンソ®配合錠切替群と、BIC/FTC/TAFの投与を継続したBIC/FTC/TAF継続群に2:1で無作為に割り付けた。
評価項目(有効性):
主要評価項目
HIV-1 RNA量≧50 copies/mLの患者の割合(48週時)(検証的解析項目)
副次評価項目
HIV-1 RNA量<50 copies/mLの患者の割合(48週時)
耐性変異の発現(48週時)
-ベースライン時のHIV-1プロウイルスDNAにM184I/V変異が認められた患者におけるHIV-1 RNA量<50 copies/mLの患者の割合(48週時)
CD4陽性T細胞数のベースラインからの平均変化量(48週時)等
探索的評価項目
ベースライン時の患者背景別にみたHIV-1 RNA量<50 copies/mLの患者の割合(48週時)等
評価項目(安全性):
主要評価項目
有害事象(48週時)
その他の重要な安全性評価項目
腎機能、空腹時脂質プロファイル(脂質低下薬服用患者を除く)、体重のベースラインからの平均変化量(それぞれ48週時)等
解析計画:
有効性解析の主要な解析集団は、治験薬を1回以上投与され、かつべースライン時のデータを有する患者と定義するFull Analysis Set(FAS)集団とした。
HIV-1 RNA量の欠測値の取扱いには、FDA Snapshot法を用いた。患者割合の群間差の95%信頼区間(CI)は、層別を伴わないMiettinen and Nurminen(MN)法に基づき算出した。
主要評価項目の群間差(イドビンソ®配合錠切替群-BIC/FTC/TAF継続群)の多重性を調整した両側95%CIの上限が4%未満の場合に、イドビンソ®配合錠切替群はBIC/FTC/TAF継続群に対して非劣性を示し、0%未満の場合に優越性を示すとした。
副次評価項目である耐性変異の発現は、各治療薬に対する遺伝子型又は表現型の耐性を示した薬剤耐性解析対象集団の例数を48週時点で投与群ごとに要約した。
Full analysis set-Resistance(FAS-R)集団には、ベースラインにプロウイルスDNA耐性データを有する被験者を含めた。
CD4陽性T細胞数のベースラインからの平均変化量の各群の95%CIはt分布に基づいて算出し、平均値の群間差及び95%CIは、各群のベースラインは同一で、投与後の各時点における各群の平均は異なり得ると仮定したcLDAモデルを用いて推定した。解析モデルには投与群、時点、時点と投与群の交互作用を含めた。
探索的評価項目として48週時のHIV-1 RNA量<50 copies/mLの患者の割合についてベースライン時の患者背景別(年齢、性別、地域、人種、民族、BIC/FTC/TAFの治療期間)にサブグループ解析を実施した。サブグループ解析の群間差の推定値(及び層別しないMN法を用いた95%CI)についてはベースライン別の分類変数で計算し、欠測値の取扱いには、FDA Snapshot法を用いた。
安全性解析対象集団は、無作為化され、治験薬が1回以上投与された患者のAll participants as treated(APaT)集団とした。
その他の重要な安全性評価項目である48週時の腎機能、脂質、体重のベースラインからの平均変化量の95%CIはt分布に基づき算出した。
主要評価項目である48週までの中間解析結果を提示するが、本試験の主要パートは144週間であり、試験は継続中である。
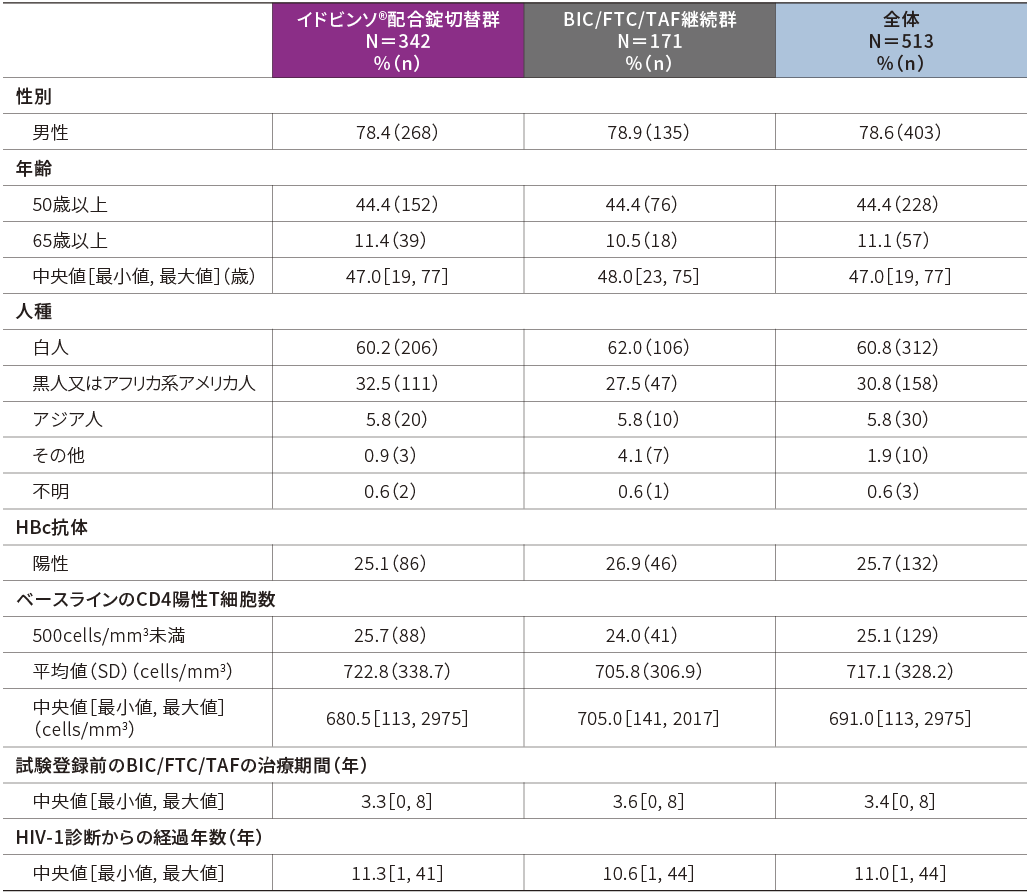
2.患者背景(FAS)
3.有効性
■ウイルス学的有効性(48週時・FAS)[主要評価項目(検証的解析結果)・副次評価項目]
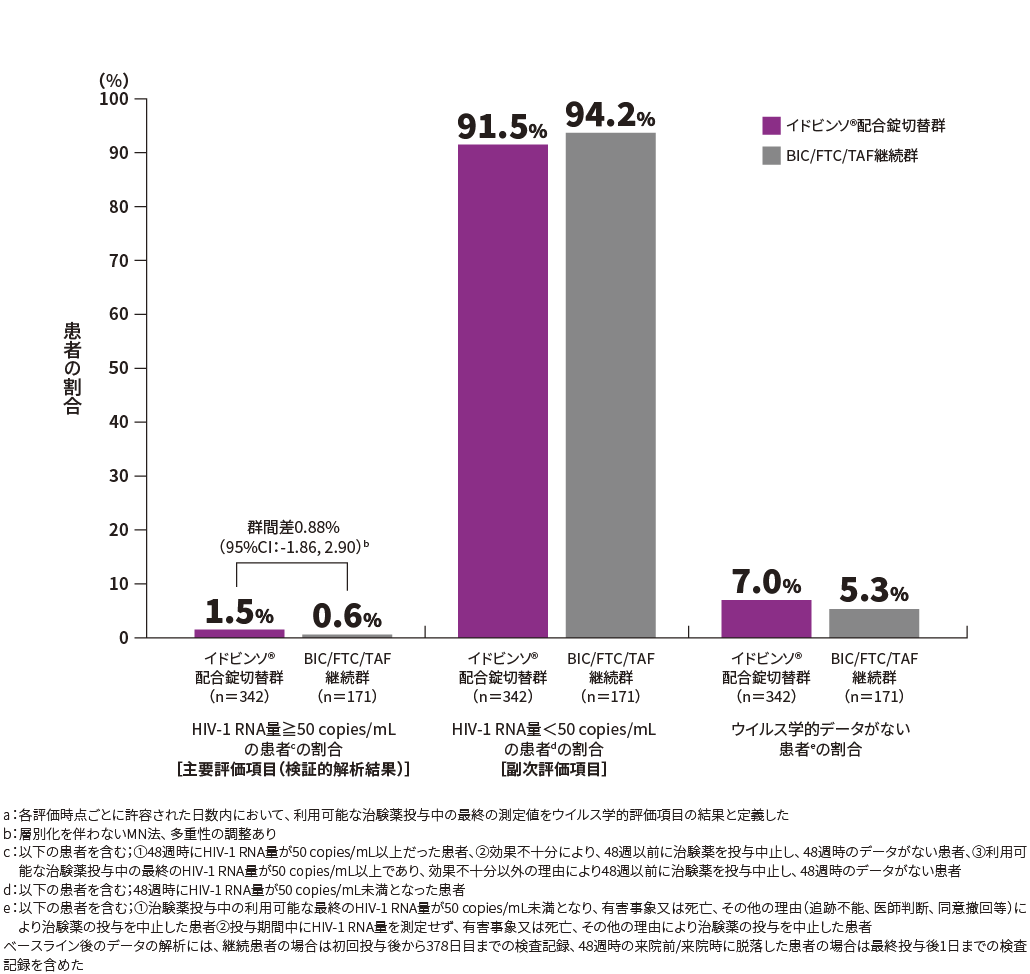
48週時におけるHIV-1 RNA量≧50 copies/mLの患者の割合は、イドビンソ®配合錠切替群1.5%(5/342例)、BIC/FTC/TAF継続群0.6%(1/171例)、群間差0.88%(95%CI*:-1.86, 2.90)であり、95%CIの上限が4%を下回ったため、イドビンソ®配合錠切替群のBIC/FTC/TAF継続群に対する非劣性が検証されました。一方、95%CIの上限が0%を下回らなかったため、優越性は示されませんでした。
*:層別化を伴わないMN法、多重性の調整あり
HIV-1 RNA量≧50 copies/mL、又は<50 copies/mLの患者の割合(48週時・FAS)[FDA Snapshot法a]
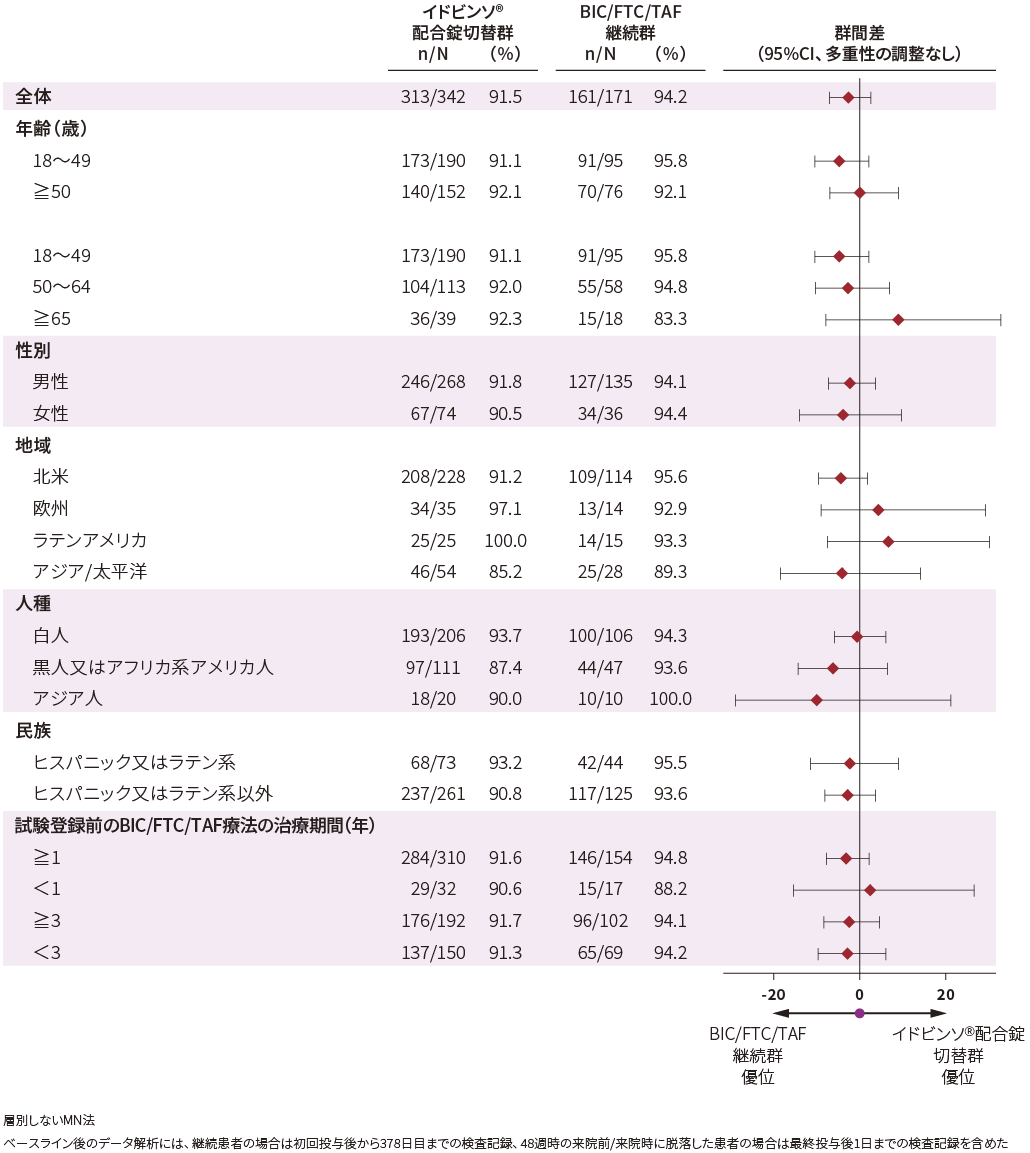
■事前規定された有効性サブグループ解析(48週時・FAS)[探索的評価項目]
ベースライン時の患者背景別にみた48週時におけるHIV-1 RNA量<50 copies/mLの患者の割合は以下の通りでした。
HIV-1 RNA量<50 copies/mLの患者の割合(48週時・FAS)[FDA Snapshot法]
■耐性変異の発現(48週時・FAS)[副次評価項目]
48週時までに、本剤に対する新耐性変異の発現は認められませんでした。

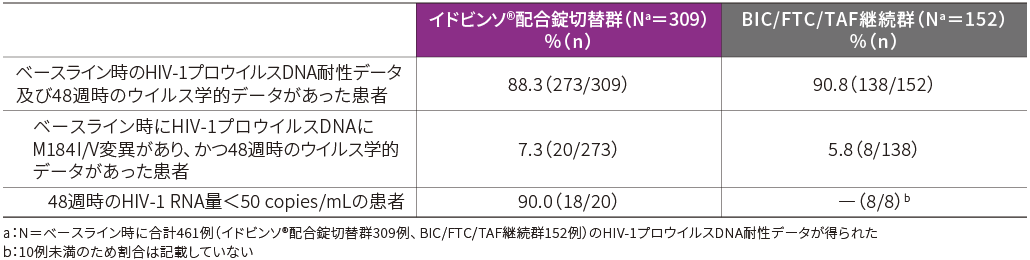
■ベースライン時のHIV-1プロウイルスDNAにM184I/V変異が認められた患者におけるウイルス学的有効性(48週時・FAS-R)[副次評価項目]
ベースライン時のHIV-1プロウイルスDNAにM184I/V変異が認められ、かつ48週時のウイルス学的データがある患者においてウイルス学的有効性が検討されました。
48週時におけるHIV-1 RNA量<50 copies/mLの患者の割合は、イドビンソ®配合錠切替群で18/20例、BIC/FTC/TAF継続群で8/8例でした。
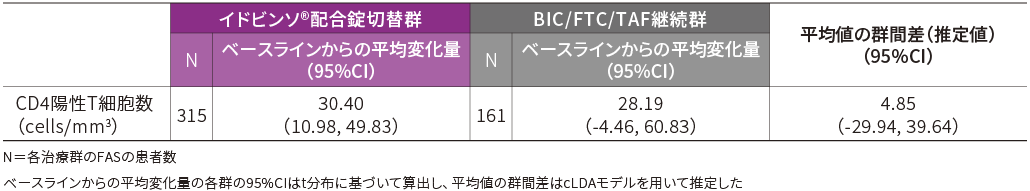
■免疫学的有効性(48週時・FAS)【副次評価項目】
48週時のCD4陽性T細胞数のベースラインからの平均変化量は以下の通りでした。
4.安全性
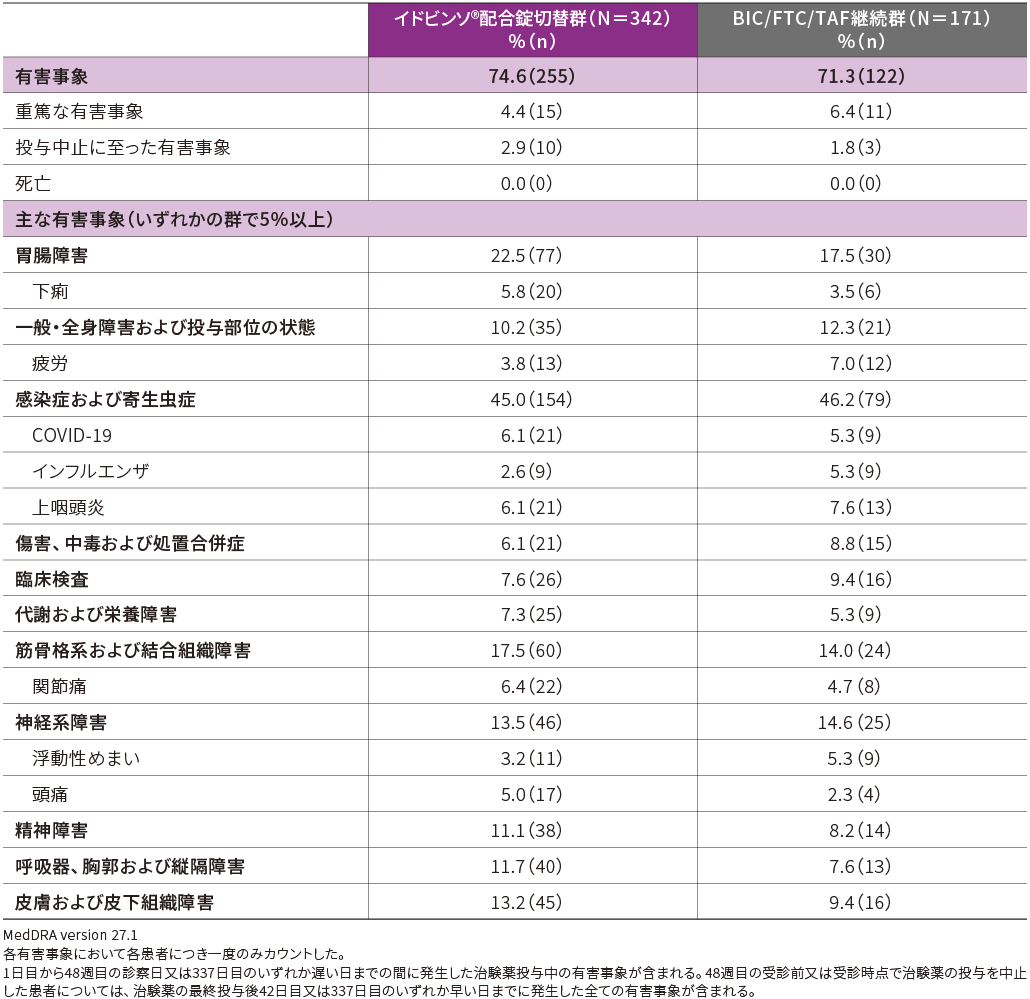
■有害事象(48週時・APaT)
主要評価項目である48週時までの有害事象発現率は、イドビンソ®配合錠切替群で74.6%(255/342例)、BIC/TAF/FTC継続群で71.3%(122/171例)でした。
主な有害事象(いずれかの群で5%以上)は、下表の通りでした。
重篤な有害事象の発現率は、イドビンソ®配合錠切替群で4.4%(15/342例)、BIC/FTC/TAF継続群で6.4%(11/171例)であり、2例以上に発現した重篤な有害事象は、イドビンソ®配合錠切替群で急性膵炎2例、BIC/FTC/TAF継続群で蜂巣炎3例でした。
投与中止に至った有害事象の発現率は、イドビンソ®配合錠切替群で2.9%(10/342例)、BIC/FTC/TAF継続群で1.8%(3/171例)であり、その内訳はイドビンソ®配合錠切替群でB型肝炎、不眠症が各2例、脱毛症、CD4リンパ球減少、リンパ球数減少、低リン血症、寝汗、免疫性血小板減少症が各1例、BIC/FTC/TAF継続群でCD4リンパ球減少、下痢が各1例、疲労及び四肢不快感が1例(同一患者)でした。
両群ともに死亡例は認められませんでした。
有害事象(薬剤との関連の有無にかかわらず)(48週時)
■副作用(48週時・APaT)
48週時までの副作用発現率はイドビンソ®配合錠切替群で10.2%(35/342例)、BIC/FTC/TAF継続群で9.4%(16/171例)でした。
主な副作用(いずれかの群で1%以上)は、イドビンソ®配合錠切替群で下痢1.5%(5/342例)、鼓腸、頭痛、そう痒症が各1.2%(4/342例)、BIC/FTC/TAF継続群で体重減少、食欲減退、異常な夢、睡眠障害が各1.2%(2/171例)でした。
重篤な副作用は、イドビンソ®配合錠切替群で免疫性血小板減少症が1例でした。
投与中止に至った副作用は、イドビンソ®配合錠切替群で不眠症、リンパ球数減少、寝汗、免疫性血小板減少症が各1例、BIC/FTC/TAF継続群で四肢不快感と疲労が1例、下痢が1例でした
両群ともに死亡は認められませんでした。
副作用(48週時)

■腎機能への影響(48週時・APaT)[その他の重要な安全性評価項目]
eGFRのベースラインからの平均変化量

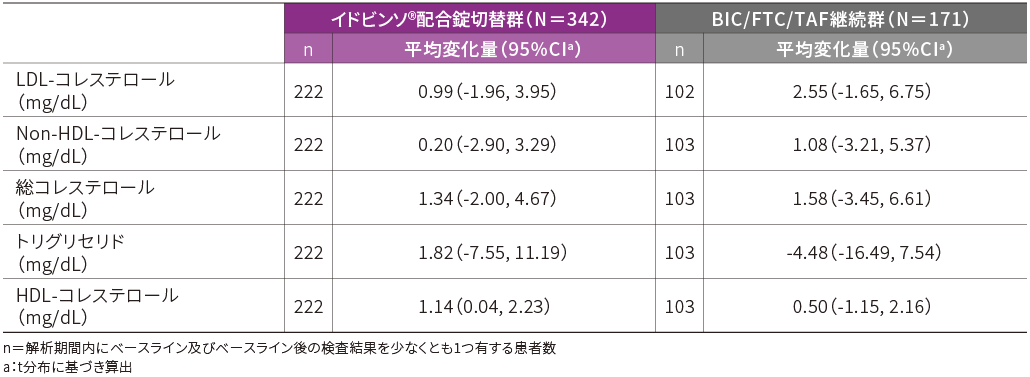
■脂質への影響(48週時・APaT)[その他の重要な安全性評価項目]
空腹時脂質プロファイルのベースラインからの平均変化量(脂質低下薬服用患者を除く)
■体重への影響(48週時・APaT)[その他の重要な安全性評価項目]
体重のべースラインからの平均変化量

関連コンテンツ

イドビンソ®配合錠の作用機序
イドビンソ®配合錠の3つの作用機序について動画でご紹介します。ご監修:国立大学法人鹿児島大学 ヒトレトロウイルス学共同研究センター 抗ウイルス療法研究分野 教授 […]
製品基本Q&A
イドビンソ®・感染症関連領域情報

5学会による新型コロナウイルス感染症 診療の指針のポイント
こちらの動画では、5学会による新型コロナウイルス感染症 診療の指針のポイントについてご紹介します。
イドビンソ®配合錠の作用機序
イドビンソ®配合錠の3つの作用機序について動画でご紹介します。ご監修:国立大学法人鹿児島大学 ヒトレトロウイルス学共同研究センター 抗ウイルス療法研究分野 教授 […]

同種造血幹細胞移植後の晩期CMV再活性化リスクとプレバイミス®によるCMV感染管理
同種造血幹細胞移植後の晩期CMV再活性化リスク、特にPTCy後のCMV感染管理についてプレバイミス®の有効性・安全性を含めて紹介しています。